
Abstract
Pancreatic ductal adenocarcinoma is one of the most aggressive malignancies, characterized by the local invasion into surrounding tissues and early metastasis to distant organs. Oncogenic mutations of the
Keywords
Introduction
Pancreatic cancer is known as one of the most aggressive adult solid tumors. About 90% of pancreatic cancers begin in the exocrine pancreas, which forms the bulk of the gland, and secretes an enzyme-rich alkaline fluid into the duodenum via the pancreatic duct. In the recent years, significant efforts have been devoted to understanding the pathogenesis of this disease with the goal of improving its early detection and patient survival. Multiple genetic abnormalities have been identified in pancreatic cancer and grouped as a core set of a 12 cellular signaling pathways altered in the majority of the pancreatic tumors.1,2 The majority of pancreatic adenocarcinomas (more than 90%) harbor gain-of-function mutations in the
Cooperation of molecular alterations in signaling pathways makes it difficult to target the transformed cell population of the pancreas. We developed a cell model system with disrupted mutant K-RAS by introducing the homologous recombination vector into MiaPaCa-2 pancreatic cancer cells. This cell model was used to evaluate the consequences of mutant K-RAS inactivation on the ability of pancreatic cancer cells to migrate, invade, and metastasize. We identified and validated the important proteins drivers of cellular survival, migration, and metastasis such as Rho-GTPase activating proteins (Rho-GAPs), RalA signaling pathway, and caveolin-1. These sensitive effectors of mutant K-RAS activity coordinate invasive potential of pancreatic cells and present the valuable targets for the future drug development.
Materials and Methods
Materials
All cell culture reagents, restriction and DNA-modifying enzymes, LipofectAMINE-2000 reagent, and specific primers used in this study were purchased from Life Technologies, Inc. K-RAS activity kit was purchased from Millipore. The peroxisome proliferator-activated receptor gamma (PPARγ) ligand, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) was purchased from Cayman Chemical and dissolved in dimethyl sulfoxide (DMSO). The specific siRNAs were purchased from Santa Cruz Biotechnology, Inc. Apoalert® Annexin V kit was purchased from Clontech Laboratories, Inc., Takara Bio Company.
Cell lines
The MiaPaCa-2 (
RNA isolation
Total RNA was extracted from MiaPaCa-2 cells, and clones were plated for 48 hours after subculture using the Qiagen RNeasy Kit as per the manufacturer's protocol. RNA samples were treated with DNase to remove residual DNA using Ambion® DNase treatment and removal kit (Ambion™ by Life Technologies, Inc., Cat.# AM1906) and evaluated for integrity of 18S and 28S rRNA by ethidium bromide staining of 1 μg of RNA resolved by electrophoresis on a 1.0% agarose/formaldehyde gel.
cDNA microarray
For cDNA microarray, samples preparation, experimental design, and analysis were performed as described previously.19–21 The cDNA chip had ~5,300 human genes, with more than 3,000 known genes and ~2,300 expressed sequence tags, as determined by UniGene. The cDNA sequences printed on the microarray were derived from cDNA clones produced by the IMAGE consortium. Hybridizations of cDNA samples of MiaPaCa-2 versus clones were performed in triplicate. Genes were considered to have altered expression levels when a mean 2-fold or greater change in their expression level and a
Quantitative real-time PCR
Total RNA was prepared as described above. The cDNA was made using Applied Biosystems High Capacity cDNA Reverse transcription kit (Part #4368814). Real-time PCR amplification was performed with an ABI PRISM 7700 SDS instrument (Applied Biosystems by Life Technologies Inc.), utilizing the universal thermal cycling conditions recommended by the Assay-on-Demand products protocol and by using Applied Biosystems High Capacity cDNA Reverse transcription kit (Part #4368814). Each 20 μL PCR reaction included 10 μL of TaqMan Universal PCR master mix, 4 μL of the resulting cDNA from the reverse transcription step, and 6 μL diluted primer and probe mixes ordered from Assay-on-Demand products (Applied Biosystems, by Life Technologies). No template controls were included in each plate to monitor potential PCR contamination. The expression of genes was tested in triplicate, and each reaction was run in duplicate. To determine the relative expression level of each gene, the comparative
For the analysis of K-RAS expression level, RNA was isolated from MiaPaca-2, M-27 and colon cell lines Caco-2 (wild type K-RAS) and HCT116 (K-RasG13D) at 48 h after subculture and converted into cDNA using High Capacity cDNA Reverse transcription kit (ABS, Part # 4368814). cDNA was amplified using
Growth curve
The MiaPaCa-2 cells were plated in six-well plates with a concentration of 0.1 × 106 cells/well. The MiaPaCa-2 parental cells and clones were plated in triplicate. The cells were grown for 24, 48, 72, and 96 hours. The cells were harvested and counted using an automated Trypan blue dye exclusion method on a Beckman Coulter Vi-Cell™ Cell Viability analyzer (Beckman Coulter Inc.). Cell size analysis was done using cell measurements obtained from Beckman Coulter Vi-Cel™ Cell Viability analyzer.
RAS activity assay
The RAS activity assay was performed with a kit from Millipore, according to the manufacturer's protocol. The MiaPaCa-2 parental cells and clones were seeded at 1.5 × 106 cells/100 mm plate. After 48 hours, the cells were scraped from the plates after adding 0.5 mL 1 × Mg2+ lysis buffer (MLB). The cell lysates were prepared to have a concentration of 500 μg/μL. Each cell lysate sample was divided into two tubes. The tubes were incubated either with 2.5 μ
Western analysis
MiaPaCa-2 parental and clonal cells were collected 48 and 96 hours after subculture by lysing on ice in radioimmunoprecipitation assay buffer (phosphate buffered saline [PBS], 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 30 μg/mL aprotinin, 100 mM sodium orthovanidate, and a 10 mg/mL phenylmethylsulfonyl fluoride). Samples were kept on ice for 30 minutes, followed by centrifugation at 13,000 ×

Apoptosis assay by Annexin V
MiaPaCa-2 parental cells and clones were seeded in 6-well plates at 0.1 × 106 cells/well. Twenty-four hours after subculture cells were treated with DMSO at the concentration of 0.05 v/v% or 0.5 v/v%. The MiaPaCa-2 parental cells and clones were treated for 48 hours and then were harvested and processed for the flow cytometry analysis according to the manufacturer's protocol (Clontech Laboratories, Inc.)
Membrane isolation
Two 100 mm plates per cell type were plated with 1 × 106 cells per plate. The cells were collected at 48 hours after subculture. The membrane extractions were done using MEM_PER™ Plus membrane Protein extraction Kit (Thermo Fisher Scientific, Cat.#89842) as per the manufacturer's protocol.
Cell migration and invasion assay
MiaPaca parental cells and

For other invasion experiments (substrate specificity, siRNAs), the stained inserts were dissolved in 200 μL 0.1 M citric acid in 96-well plate while shaking for 5 minutes on a high speed plate shaker. The supernatant was then transferred to a new well and read at 560 nm on an EL800 Universal Microplate Reader (Bio-Tek Instruments, Inc.). Cell invasion assays to test substrate specificity were performed using BIOCOAT® control cell culture inserts (BD Biosciences Discovery Labware). Inserts were coated with 2 μg/well of laminin or collagen IV (Sigma-Aldrich, Life Science Research). All migration and invasion assays were carried out in sextuplet.
siRNAs transfections experiments
MiaPaCa-2 cells were seeded in Opti-MEM media supplemented with 10% FBS at a concentration of 0.3 × 106 cells/mL. Twenty-four hours after subculture, the cells were transfected with either caveolin-1 (CAV-1), siRNA (Cat.# sc-29241) or RalA siRNA (sc-41842), and control siRNA (sc-37007) (Santa Cruz Biotechnology, Inc.) at the concentration of 75 nM using DramaFECT Duo transfection reagent (Dharmacon™; GE Healthcare, Inc.), according to the manufacture's protocol. Twenty-four hours after transfection, the cells were trypsinized, counted, and processed for migration and invasion assays as described earlier. All siRNA transfections were done in triplicates, following by migration and invasion assays in sextuplet for each siRNA and experiments were repeated twice. Statistical analysis was done in each experiment independently.
F-actin visualization
MiaPaCa-2 cells and M-27 clone were seeded on chamber slides (Lab-Tek; Nalge Nunc International) at concentration 1 × 105 cells/chamber. Cells were grown for 96 hours after subculture before staining. Immunofluorescence staining for F-actin was carried out using ActinGreen™ 488 ReadyProbes™ reagent according to the manufacturer's instructions. Briefly, the cells were washed with PBS, fixed in fixative solution (4% formaldehyde in PBS) for 10 minutes at room temperature, permeabilized with permeabilization buffer (0.5% Triton X-100 in PBS) for 5 minutes at room temperature, washed twice with PBS, and stained with ActinGreen 4888 ReadyProbes reagent for 30 minutes. Cells were photographed with Nikon E800 confocal microscope with epifluorescence and DIC capabilities at 60× magnification.
Subcutaneous model
For the tumorigenesis study, the MiaPaCa-2 parental cell line and the three mutant
Orthotopic model
For animal survival study, MiaPaCa-2 parental and M-27 clonal cells at the concentration of 3 × 106 cells in 50 μL Matrigel (Becton Dickenson) were injected into the tail of the pancreas of anesthetized mice by using a 30-gauge needle. Animals were observed twice weekly for adverse signs associated with tumor growth (eg, >20% body weight loss, immobility, loss of grooming), at which time mice were euthanized. Metastatic colonization and tumors formed were counted postmortem.
Statistics
Statistical analysis of cell culture experimental data was performed using paired
Results
Characterization of MiaPaCa-2 pancreatic cancer cells with disrupted mutant K-RAS oncogene
RAS activity
The MiaPaCa-2 pancreatic cancer cells used in this study have characteristics found in more than 90% of human pancreatic adenocarcinomas. MiaPaCa-2 cells exhibit mutation in the
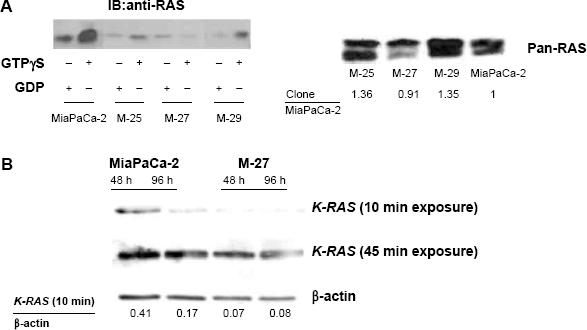
Conformation of mutant
The means, standard error of slope, and
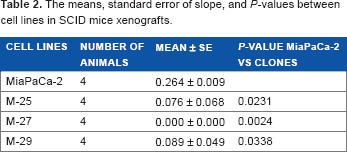
Effect of K-RAS disruption on cell growth and cell size
A statistically significant decrease in growth rates was observed in all selected clones with decreased RAS activity compared to MiaPaCa-2 cells (Fig. 2A). Additionally, the difference in growth rates among clones was noted, that is, M-27 clone grew slower than the M-25 and M-29 clones. Western blot analysis of proteins that control cell growth and proliferation showed that clone M-27 had the lowest expression levels of proliferation-specific nuclear antigen (PCNA) protein, which is involved in the control of eukaryotic DNA replication and c-MYC oncogene, controlling variety of cellular functions from transcription to metabolism (Fig. 2B). Analysis of cell parameters revealed that M-27 clone had the significantly smaller cells size compared to MiaPaCa-2 cells and the other two selected clones: M-25 and M-27 (Fig. 2C and Supplementary Fig. 2). This observation is consistent with the morphological alterations reported in HCT116 colon cancer cells with disrupted mutant
Growth properties of MiaPaCa-2 and clones with disrupted mutant
Apoptosis
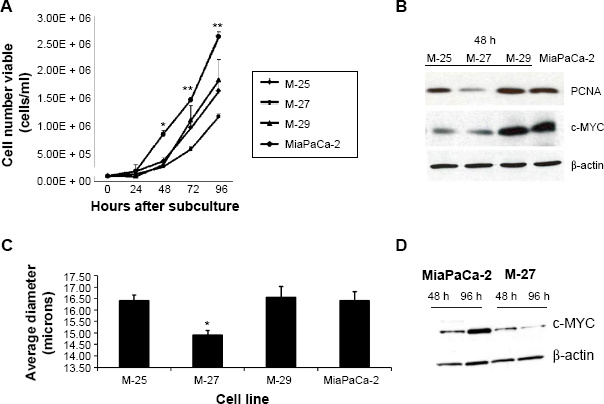
Suppression of apoptosis is one of the important and well-documented consequences of mutant K-RAS expression in cancer. 23 It has been known that activated K-RAS can inhibit apoptotic signaling cascade through its effector PI3K. PI3K is in turn activates AKT and leads to suppression of apoptosis via several mechanisms, such as inhibition of caspases activation and inactivation of proapototic Bcl-2 family proteins. Analysis of downstream transducers of activated K-RAS in MiaPaCa-2 showed that expression of phosphorylated PI3, ERK, and AKT kinases in M-27 clone was lower at 96 hours after subculture (Fig. 3A). This indicates that inactivation of RAS in M-27 clone altered multiple cellular processes, including apoptosis signaling pathway. Different natural and synthetic compounds can induce the cell death through apoptosis. We have tested the effect of PPARγ ligand, 15d-PGJ2, on apoptosis induction in MiaPaCa-2 clones with disrupted mutant K-RAS. 15d-PGJ2 is a natural derivative of prostaglandin D2, a member of the cyclopentenone family, and has been reported previously to inhibit the pancreatic cell growth and invasiveness and induce apoptosis through activation of caspace-8, 9 and 3.24,25 We found that disruption of mutant K-RAS increased the cells sensitivity to 15d-PGJ2 treatment, such as we observed the induction of cleaved caspase 3 expression in MiaPaCa-2 clones, but not in MiaPaCa-2 parental cells upon treatment with 15d-PGJ2 at the concentration of 1 μM (Fig. 3B). As has been previously reported, the optimal concentration of 15d-PGJ2 necessary for the induction of apoptosis in MiaPaCa-2 parental cells is 35 μM. 25
Mutant
We also observed the increased number of apoptotic and necrotic cells in clones M-25 and M-27 when DMSO was added to the cultured media at the concentration of 0.5 v/v% (Fig. 3C).
Migration and invasion
We further evaluated the ability of MiaPaCa-2 clones to migrate and digest extracellular matrix proteins using cell migration and Matrigel invasion assay. MiaPaCa-2 parental cell line with the activated mutant
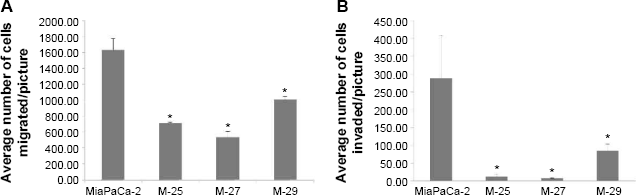
Interaction of MiaPaCa-2 cells and clones with basement membrane in vitro by Matrigel invasion assay. (
SCID mouse analysis
We further tested the ability of MiaPaCa-2 to form xenograft and orthotopic tumors in SCID mice. In xenograft study, all clones showed the significant delay in tumor growth compared to MiaPaCa-2 parental cell line (Fig. 5A and Table 2). In particular, the M-27 clone had the increased latency of tumor development (no palpable tumors 62 days postinjections when the experiment was terminated). Following this study, we tested the ability of M-27 clone to form metastasis using orthotopic mouse model. After the injection of MiaPaCa-2 parental cells and M-27 clone, embedded in Matrigel, into the tail of the pancreas, SCID mice were monitored weekly, sacrificed as they become morbid, and analyzed for the presence and location of metastasis. The duration of this experiment was 57 days. The Kaplan–Meier survival curves showed the statistically significant increase in survival rates of animals injected with M-27 clone (average survival time 48 days MiaPaCa-2-injected animals versus 53 days for M-27 clone-injected animals, Fig. 5B,
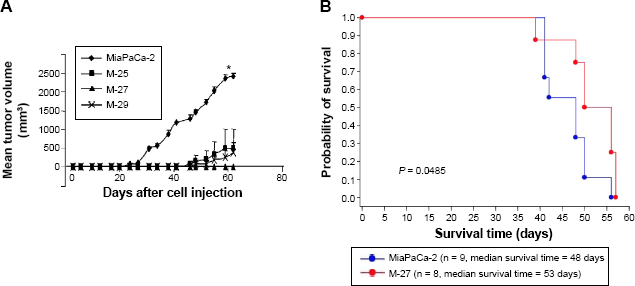
Analysis of tumorigenicity of MiaPaCa-2 cells and clones in vivo. (
The statistical significance of difference in the incidence of metastases between the groups.

Molecular profiling of the MiaPaCa-2
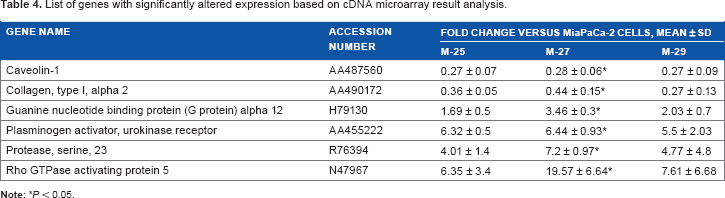
The genetic and functional assays described earlier allowed us to conclude that inactivation of mutant
List of genes with significantly altered expression based on cDNA microarray result analysis.
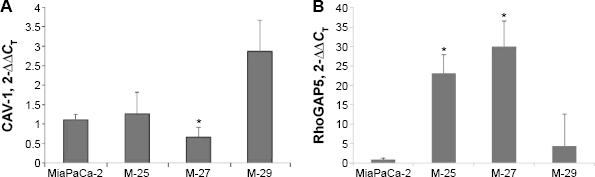
Validation of altered expression of RhoGTPase 5 (
Functional validation of mutant K-RAS induced alterations in MiaPaCa cell culture model
We further evaluated the downstream effectors of RAS signaling in M-27 clone, which are regulated by CAV-1 and
Caveolin-1
We identified and confirmed decrease in CAV-1 expression both at the mRNA (Fig. 6A) and protein (Fig. 7A) levels. We further performed membrane and cytosolic fractionations to determine CAV-1 distribution within cells. CAV-1 is mainly localized in the membrane fraction, and its phosphorylated form is found in the cytosol fraction (Fig. 7B). Expression of CAV-1 and its phosphorylated form was reduced in both the cytosol and in membrane fractions, of M-27 clone by approximately 2-fold (as measured by ImageJ software) compared to MiaPaCa-2 cells, which is consistent with observed downregulation of CAV-1 transcript (Fig. 7A). To demonstrate the direct role of CAV-1 in pancreatic cell migration, we transiently transfected siRNA for CAV-1 into MiaPaCa-2 cells and measured the ability of these cells to migrate and invade. The knockdown of CAV-1 was confirmed by Western blot analysis in cells transfected with siRNA specific to CAV-1 (Fig. 7C, upper panel). Cells were depleted of CAV-1 protein and cell migration and cell invasion were significantly suppressed (
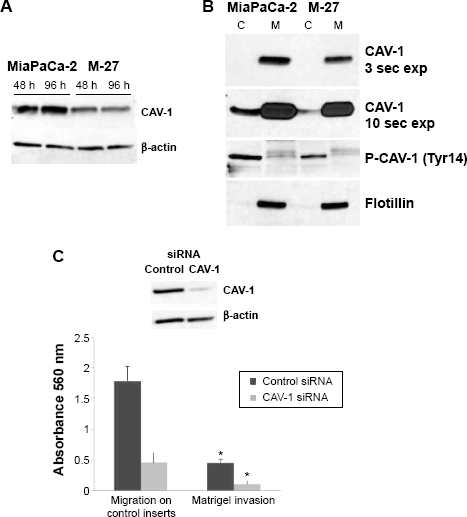
Caveolin-1 downregulation in pancreatic cells with disrupted mutant K-RAS signaling causes decrease in pancreatic cells migration and invasion. (
RhoGAP 5
Another transcriptional target identified in M-27 cells was
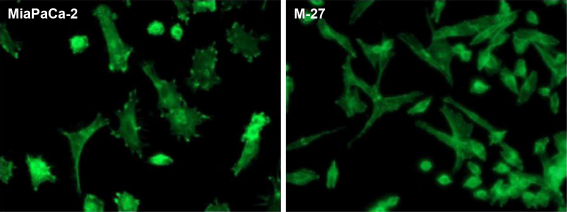
Representative immunofluorescence images of F-actin staining in MiaPaCa-2 and M-27 cells at 96 hours after subculture. Samples were collected and processed as described in “Materials and methods” section.
RalA GTPase
Another RAS-controlled pathway that can diverge to F-actin assembly is the RalA GTPase signaling pathway. 28 RalA and RalB are members of the RAS family of small G proteins that are activated downstream of Ras via RalGEFs. Both RalA and RalB share approximately 80% homology at the amino acid level, but they have distinct roles in normal cells and during tumorigenesis. 29 We found a twofold decrease in the level of RalA protein in M-27 clone compared to MiaPaCa-2 cells. In contrast to RalA, the level of RalB protein in M27 cells was similar to the one observed in MiaPaCa-2 cells (Fig. 9A). The level of Ral binding protein 1 (RalBP1) downstream of RalB did not change (Fig. 9A). Using siRNA for RalA, we found that RalA knockdown (Fig. 9B, upper panel) does not affect MiaPaCa-2 cell migration but significantly inhibits cell invasion (Fig. 9B, lower panel). This observation is consistent with finding reported in human bladder cancer cells. 29 One last observation we would like to report is the significant suppression of MET/hepatocyte growth-factor (HGF) receptor tyrosine kinase (MET RTK) in M-27 clone compared to MiaPaCa-2 cells, while the level of CD44 protein adaptor was similar in both cell lines (Supplementary Fig. 4). Although RAS usually acts downstream of RTKs, RAS disruption in our system may indirectly influence MET production through adapter proteins such as growth-factor-receptor-bound protein 2 (GRB2) and GEF Son of Sevenless (Sos).
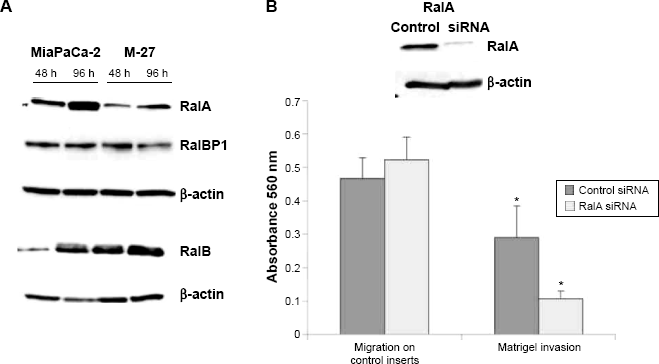
RalA inhibition suppresses cell invasion in MiaPaCa-2 pancreatic cells. (
Discussion
There are numerous attempts underway to target different molecular pathways in pancreatic cancer with the goal to combat this disease. Unfortunately, the standard treatments for advance disease are mostly ineffective.
30
K-RAS is the membrane-associated guanine nucleotide binding proteins of ~21 kDa that acts as a molecular switch for signal cascades that modulate many aspects of cell behavior, including proliferation, differentiation, motility, and death.
31
Due to alternative exon 4 utilization of K-RAS gene, there are two splice variants, KRAS4A and KRAS4B, expressed in eukaryotic species, with K-RAS4B isoform being the most prevalent in human cells. Constitutive activation of
Activating
The novelty of current research is two-fold: first, we attempted to mimic pancreatic tumor variability by using homologous recombination approach to disrupt mutant K-RAS activity. We identified and characterized three MiaPaCa-2 clonally selected cell lines with the variable mutant K-RAS activity. Second, we focused on identification of the drugable downstream K-RAS effectors controlling pancreatic tumors invasion and metastasis.
We developed clonally selected pancreatic cell lines, which express homologous recombination vector targeting K-RAS oncogene. The homologous recombination-based gene targeting technology requires homologous recombination efficiency of the cell line and usually results in a high proportion of nontargeted random integration. This is due to the significantly higher genomic insertion efficiency compared to homologous recombination efficiency. 39 Following the positive/negative selection approach we screened 35 clonally selected recombinants and identified clone M-27 out of 35 screened clonally selected recombinants as the best targeted clone. We used clone M-27 alone with two other clones, M-25 and M-29, which exhibited the variable degree of suppression of mutant K-RAS activity, for further evaluation.
We found that the PI3K/AKT pathway was the main RAS effector that transduces RAS signaling in MiaPaCa-2 cells and regulates apoptosis, migration, and invasion. MiaPaCa-2 clones with low mutant K-RAS activity showed the higher sensitivity to apoptosis-inducing agent, 15-deoxy[delta]12,14-prostaglandin J2 (15d-PGJ2). This finding confirms that AKT inhibitors, which are currently under active investigation as single agents or their combinations with apoptosis-inducing agents, is a promising approach for restriction of pancreatic cancer progression. 40
We showed that MiaPaCa-2 clone phenotypes depend on their level of RAS activity. Clone M-29 is most similar to the MiaPaCa-2 parental cells in cell growth and size characteristics and migration and invasion in vitro, whereas M-27 clone, which expressed the lowest level of activated RAS, had the lowest number of cells that were capable of migration and invasion through Matrigel.
We tested the consequences of mutant
For comparison with observations found in MiaPaCa-2 clones, we used the homologous recombination technique with pBKNT-D-targeted vector to disrupt
By comparing the gene expression profiles of MiaPaCa-2 parental cell line and clones, we have identified and validated two genes,
RAS superfamily of proteins is regulated via intrintic guanine nucleotide exchange and GTP hydrolysis. We found that the specific regulator of Rho family of GTPase, RhoGAP5, was upregulated in M-27 clone at the transcriptional level. The posttranslational regulation of RhoGAP5 by Src family tyrosine kinases also has been reported. 41 Rho GTPases act as molecular switches to control signal transduction pathways by cycling between an inactive, GDP-bound and an active, GTP-bound form. The GTP-bound Rho GTPases interact with a variety of downstream effectors to regulate different intracellular processes including cell growth and differentiation and cell migration. 42 In their function in migration and invasion, Rho GTPases mediate changes in actin dynamics during the cell cycle and in the specialized cell structures (lamellipodium, podosomes and focal adhesions). 43 In this regard, inhibition of RhoGTPase activity by the RhoGAPs counteracts tumor growth and invasion, and therefore, RhoGAPs are considered as tumor suppressors.44,45 There attempts are underway to evaluate RhoGTPase and RhoGAPs as targets for cancer therapy. 46
We further looked at RAS-RalGTPase signaling pathway because RalGTPases play important roles in promoting the pancreatic neoplasia. Specifically, RalA GTPase has been critical to tumor initiation and RalB GTPase is involved in tumor metastasis. 47
We found that RalA, but not RalB, protein expression is suppressed in M-27 clone with disrupted mutant K-RAS. We showed that inhibition of RalA expression with siRNA leads to suppression of MiaPaCa-2 cell invasion, but not migration. Altered Ral GTPases expression was not noted by the cDNA microarray screening. But the recent study by Gentry et al provided the evidence for the role of posttranslational modification in Ral protein functions initiated by the carboxyl-terminal hypervariable regions carboxyl-terminal tetrapeptide CAAX-motif. Both RalA and RalB require RAS converting enzyme CAAX endopeptidase for association with plasma membrane. These posttranslational modifications play important role in stability and function of RaL GTPases. 48 Therefore, the RalGEF-Ral signaling pathway is an excellent candidate for future design of therapeutic interventions in pancreatic cancer.49–51
Caveolin-1 is a major component of membrane caveolae. It is an important regulator of signaling and molecule trafficking, as well as a mediator of cell proliferation and metastasis.
52
We found that CAV-1 transcription is downregulated in MiaPaCa-2 with disrupted mutant K-RAS. Using siRNA approach, we confirmed that downregulation of
Lastly, our finding of downregulation of the MET/ HGF receptor tyrosine kinase in M-27 clone indicates that this is another candidate for future interventions. RTK are involved in cell-cell communication, metabolism, and cell motility. 54 MET is overexpressed in up to 80% of invasive pancreatic cancers and correlates with poor overall patient survival. 55 It has been reported that MET knockdown reduces tumor burden in orthotopic pancreatic cancer model. 56 The RTK signaling pathway is upregulated in pancreatic cancer, and efforts have been put into preclinical and clinical development of antagonists of HGF/MET with several Phase II clinical trials. 57
Despite the significant contribution of molecular research into pathogenesis of pancreatic cancer, the aggressive nature of this disease is not completely understood. Our findings open the possibilities of combined targeting AKT, RalA, and possible MET, pathways with different small molecule inhibitors (Fig. 10) to amplify the effect of anti-RAS therapy in metastatic pancreatic cancer.
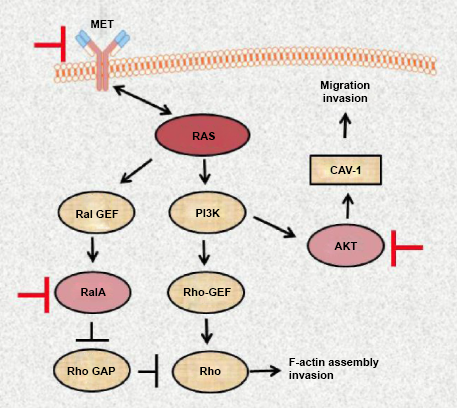
Diagram indicating the drugable targets (MET, RalA, and AKT) of RAS signaling pathways based on changes observed in MiaPaCa-2 pancreatic cell line after disruption of mutant
Author Contributions
Conceived and designed the experiments: NAI. Analyzed the data: JP, RSH, HChen, BAS, HCui, NAI. Wrote the first draft of the manuscript: JP. Contributed to the writing of the manuscript: RSH, HChen, BAS, HCui. Agree with manuscript results and conclusions: All. Jointly developed the structure and arguments for the paper: JP, RSH, NAI. Made critical revisions and approved final version: JP, RSH, NAI. All authors reviewed and approved the final manuscript.
Footnotes
Abbreviations
Acknowledgments
We thank Dr. Senia Shirasawa and Dr. Takehiko Sasazuki, Japan Central Research Institute for Advance Molecular Medicine, Tokyo, Japan for providing the vector construct for targeted disruption of the human