
Abstract
It is evident that components of the extracellular matrix (ECM) act as danger-associated molecular patterns (DAMPs) through direct interactions with pattern recognition receptors (PRRs) including Toll-like receptors (TLRs) and inflammasomes. Through these interactions, ECM-derived DAMPs autonomously trigger sterile inflammation or prolong pathogen-induced responses through the production of proinflammatory mediators and the recruitment of leukocytes to sites of injury and infection. Recent research, however, suggests that ECM-derived DAMPs are additionally involved in the resolution and fine-tuning of inflammation by orchestrating the production of anti-inflammatory mediators that are required for the resolution of tissue inflammation and the transition to acquired immunity. Thus, in this review, we discuss the current knowledge of the interplay between ECM-derived DAMPs and the innate immune signaling pathways that are activated to provide temporal control of innate immunity.
Keywords
Introduction
There is growing evidence that components of the extracellular matrix (ECM) are crucial regulators of tissue inflammation and innate immunity.1–4 Inflammation is an essential mechanism through which the innate immune system initiates a protective response to pathogen invasion and tissue injury.5,6 The activation of innate immunity results from the recognition of specific molecules by pattern recognition receptors (PRRs) that are present in both immune and non-immune cells. 7 Activation of PRRs initiates an inflammatory cascade consisting of cytokine transcription and secretion, recruitment of leukocytes into the site of infection or injury and, in the case of tissue infection, the transition to adaptive immune response which is often required for the clearance of a pathogen. 8 Activation of PRRs involves the engagement of various co-receptors and promotes specific intracellular signaling cascades upstream of the secretion of proinflammatory mediators. 9 Fine-control of the innate immune system is critical because this response must switch from the production of proinflammatory mediators to those mediators required for the resolution of tissue inflammation and the transition to an adaptive immune response. The inability to make this switch will result in increased tissue injury, chronic inflammation, and fibrosis.
PRRs recognize two classes of specific molecules resulting either from the presence of pathogens, via pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), or from danger-associated molecular patterns (DAMPs), which are released from damaged tissue. 9 While PAMPs are external agents derived from viruses or bacteria, DAMPs represent endogenous sterile stimuli, which are released either from the ECM (e.g., proteolytically digested ECM fragments) or from dying cells (e.g., calcium-binding proteins, ATP, chromatin, DNA, nuclear proteins).10,11
A broad spectrum of DAMPs has now been extensively described to promote a host of diverse biological processes involving either homeostasis and regeneration or development of pathological conditions, such as chronic inflammation and fibrogenesis.1,5,11–15 Intracellular DAMPs refer to immunogenic molecules that are released from the breakdown of necrotic and apoptotic cells or cells that undergo autophagy. 5 Such molecules are mitochondrial DNA (mtDNA) and ATP that originate from mitochondria, 16 calcium-binding protein S-100, 17 myosin fragments, 18 actin, 19 heat shock proteins, 20 uric acid 21 from cytosol, high-mobility group protein B1 (HMGB1) from the nucleus or autophagosome, 22 and histone from the nucleus 23 (Table 1). A number of thorough review articles cover our understanding of the inflammatory signaling pathways triggered by these ligands.5,14,24,25
The Nature and Source of Intra- and Extracellular DAMPs.
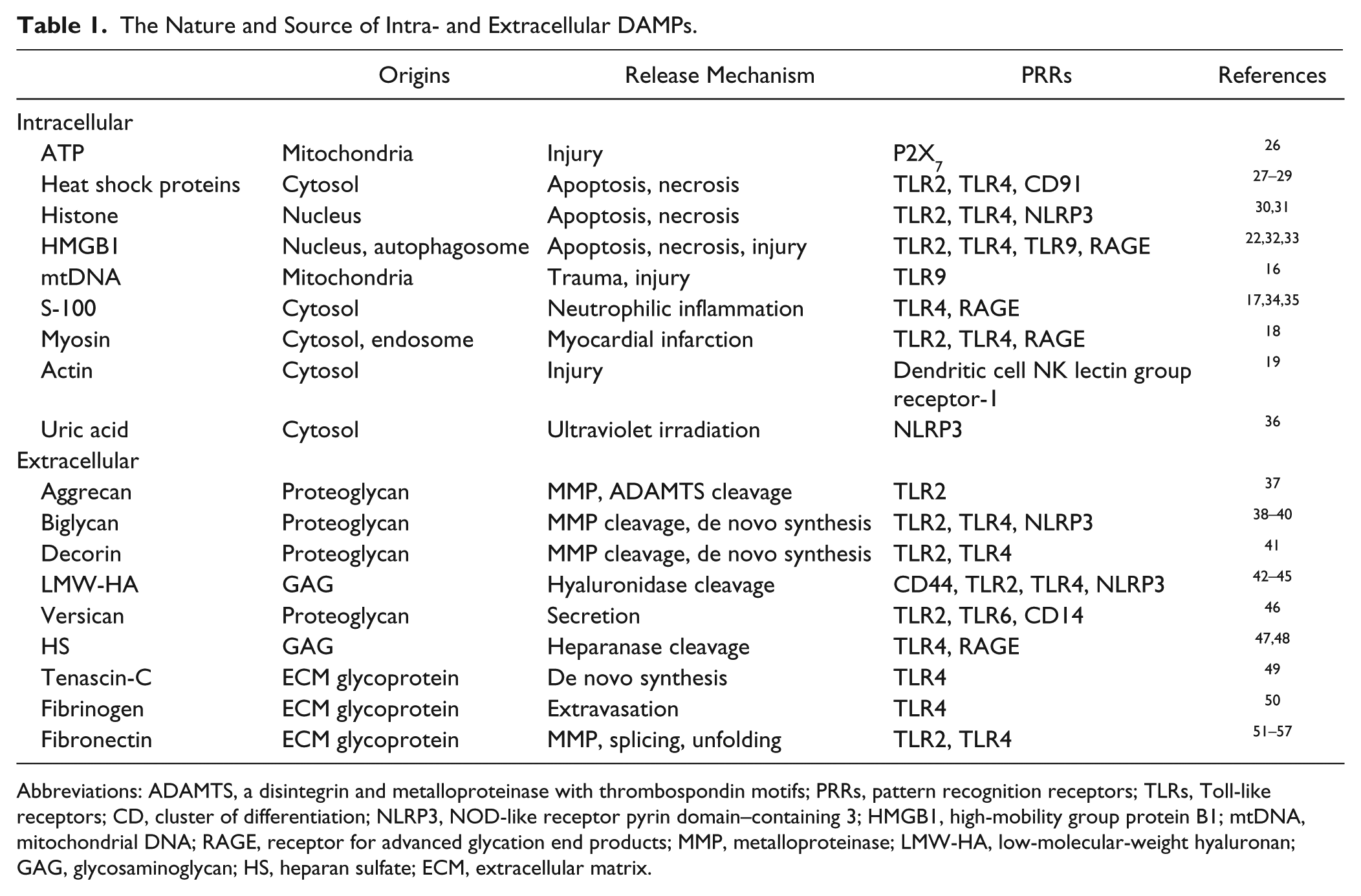
Abbreviations: ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; PRRs, pattern recognition receptors; TLRs, Toll-like receptors; CD, cluster of differentiation; NLRP3, NOD-like receptor pyrin domain–containing 3; HMGB1, high-mobility group protein B1; mtDNA, mitochondrial DNA; RAGE, receptor for advanced glycation end products; MMP, metalloproteinase; LMW-HA, low-molecular-weight hyaluronan; GAG, glycosaminoglycan; HS, heparan sulfate; ECM, extracellular matrix.
The ECM components discovered to be capable of acting as DAMPs are glycoproteins (e.g., fibrinogen, fibronectin [FN] domains, tenascin-C),50–52,58,59 proteoglycans (PGs; for example, small leucine-rich proteoglycans [SLRPs], versican),38,41–43,60,61 or glycosaminoglycans (GAGs; for example, low-molecular-weight hyaluronan [LMW-HA] and HS).47,62,63 In addition the matrix-derived protein matrilin-2 in its soluble form promotes inflammation and axonal damage 64 (Table 1).
The ECM-derived DAMPs are autonomous triggers of the inflammatory process through direct interaction with the specific PRRs. They are also capable of ramping-up and prolonging pathogen-induced inflammatory responses. 65 At later stages, depending on the pathology and signaling pathway involved, DAMPs can act to maintain or reduce the inflammatory response. To this end, DAMPs indirectly influence the innate immune response by modulating the production or activity of other sterile stimuli such as transforming growth factor (TGF)-β and interleukin (IL)-1β, which in turn will regulate the immune response.5,7,66
In this context, the present review aims to focus on the current knowledge regarding the direct and indirect interplay between ECM-derived DAMPs and the innate immune signaling pathways.
ECM-Derived DAMP/Receptor Interactions and Signaling
Several innate immune receptors have been shown to represent the first line of defense following direct interaction with danger molecules. 67 These innate immune responses are initiated by the activation of PRRs following ligand recognition and further promotion of signaling cascades. 68 DAMPs are typically released from inside the cell (e.g., heat shock proteins) or the ECM network (e.g., biglycan, decorin). Upon tissue injury, they can be released in the extracellular milieu and further trigger the innate response via PRRs either aggravating inflammation or promoting the regeneration of injured tissue.1,5,69 ECM-derived DAMPs can be generated as a consequence of enzymatic cleavage, de novo synthesis by macrophages and/or stromal cells60,70–72 as well as splicing mechanisms 53 and unfolding due to mechanical contractility. 54
To a large extent, the outcome depends on the nature of the respective DAMP, the signaling receptor, and the signaling pathway involved. 73 Common receptors for DAMPs include Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), receptor for advanced glycation end products (RAGE; as well as scavenger receptors), 74 integrins, 75 and cluster of differentiation 44 (CD44). 44 Given the large number and diversity of immune receptors, in this section, we will only address those receptors, which directly interact with ECM-derived DAMPs in relation to their consequent signaling pathway. The common structural and functional features of these immune receptors are covered by a number of reviews in great detail.10,67,76–83
TLR-Dependent ECM-Derived DAMP Signaling
TLRs are the most well-characterized PRRs, which consist of 10 distinct proteins in humans (TLR1-10) and 12 in mice (TLR1–TLR9, TLR11–TLR13; TLR10 is a pseudogene).84–87 Structurally, TLRs have a highly glycosylated N-terminal ectodomain with leucine-rich repeats (LRRs), responsible for ligand recognition, 88 and a cytoplasmic region with a Toll/IL-1 receptor (TIR) domain for signaling. 89 TLR1, 2, 4, 5, and 6 are located at the cell surface, and they sense various extracellular danger molecules, such as soluble ECM-derived ligands and pathogens, 2 while TLR3, 7, 8, and 9 are present at the endolysosomal membrane. 90 The latter category of receptors recognizes nucleic acids and PAMPs that undergo endocytosis to allow degradation by endosomes or lysosomes. 90 TLR10, the only TLR without identified ligand specificity, was initially described as an anti-inflammatory PRR, which is capable of attenuating the TLR2 response. 91 More recent studies, however, showed involvement of TLR10 in the signaling of ligands such as FSL-1, LPS, and flagellin. Thus, the TLR10 knockdown in monocytic cell lines diminished the production of IL-8, IL-1β, and chemokine (C–C motif) ligand (CCL) 20 in response to these ligands. 92
TLRs require co-receptors or accessory molecules for ligand recognition and further signaling activation. 93 Moreover, TLR signaling interacts with a variety of other signaling pathways making the design of new specific therapeutic agents increasingly difficult.65,94 Specific downstream effects depend on the specific DAMP, type of TLR, involvement of accessory molecules, cell/tissue type, and the overall biological context. For example, TLR4 engages cluster of differentiation 14 (CD14) and lymphocyte antigen 96 (MD2) to generate the LPS-induced inflammatory signal.95,96 On the contrary, TLR2 is capable of associating with TLR1, TLR6, CD14, and CD36; binding to integrins; and recognizing peptidoglycans and lipopeptides derived from gram-positive bacteria or mycoplasma.97,98 In addition, endosomal TLRs co-localize with CD14, which appears to be pivotal for TLR7 and TLR9 signaling. 99
The complexity of the TLR signaling requires the development of more specific therapeutic strategies to target certain ligand/receptor interactions and adaptor molecules rather than the simple neutralization of a receptor.
ECM-Derived DAMPs
ECM-derived DAMPs are primarily created by fragments of the ECM that are cleaved off during tissue injury and engage with multiple PRRs to initiate the proinflammatory response. This interaction is summarized in Table 1 and Fig. 1. In addition to this mode of generation, ECM-derived DAMPs can also be synthesized de novo by cytokine-activated macrophages and resident cells to bind PRRs and sustain their proinflammatory status. 100 Even though there are many distinct TLRs, it is of note that to date only TLR2 and TLR4 have been observed to recognize ECM-generated DAMPs.
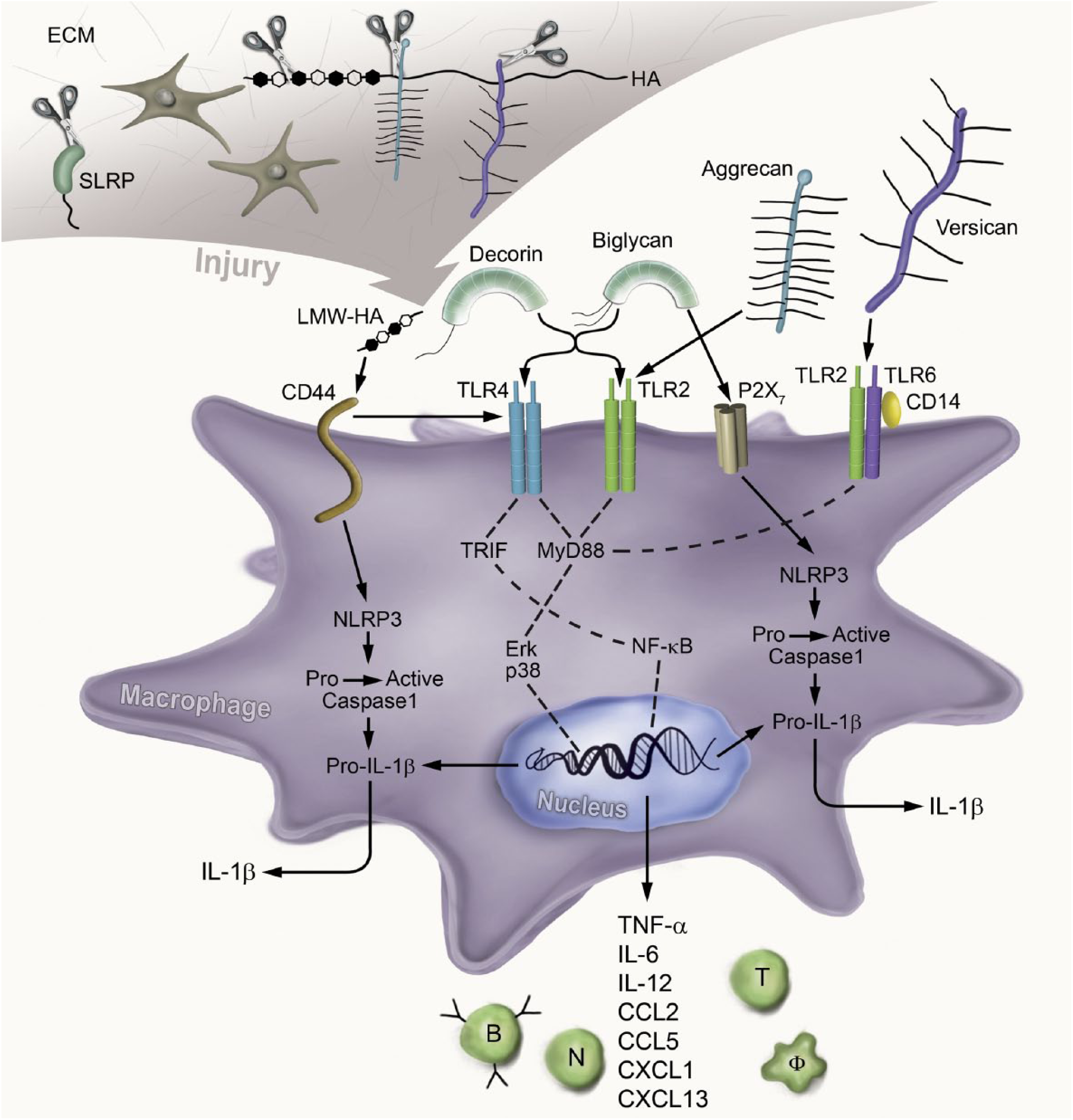
ECM-derived DAMP signaling through the innate immunity receptors. Upon injury, the ECM components HS-binding proteoglycans and SLRPs are cleaved by proteinases and the resulting soluble molecules act as DAMPs. They activate specific receptors on the surface of macrophages or intracellularly and promote inflammation. The soluble SLRPs biglycan and decorin bind to TLR2 and TLR4, aggrecan fragments signal through TLR2, and versican via the TLR2/TLR6/CD14 complex. LMW-HA signals through CD44 and further promotes the TLR4-dependent inflammatory response. These interactions promote downstream signaling, which involves the engagement of MyD88 or TRIF; the activation of NF-kB and p38, ERK MAPKs; and the expression of the proinflammatory cytokines and chemokines. Consequently, these mediators attract neutrophils, macrophages, B cells, and T cells to the site of injury. In addition, soluble biglycan clusters TLR2/4 with P2X7 and modulates the NLRP3 inflammasome. On the contrary, LMW-HA activates the NLPR3 inflammasome via CD44. Thus, biglycan and LMW-HA activate caspase-1, which cleaves pro-IL-1β into mature IL-1β. Abbreviations: ECM, extracellular matrix; DAMPs, danger-associated molecular patterns; HS, heparan sulfate; SLRP, small leucine-rich proteoglycan; TLR, Toll-like receptor; CD, cluster of differentiation; LMW-HA, low-molecular-weight hyaluronan; MyD88, myeloid differentiation primary response gene 88; TRIF, TIR-domain–containing adaptor-inducing interferon-β; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; ERK, extracellular signal–regulated kinase; MAPKs, mitogen-activated protein kinases; NLRP3, NOD-like receptor pyrin domain–containing 3; IL, interleukin; HA, hyaluronan; TNF, tumor necrosis factor; CCL, chemokine (C–C motif) ligand; CXCL, chemokine (C–X–C motif) ligand.
Biglycan and decorin, two members of the SLRPs family, generate a sterile proinflammatory response in macrophages following direct interaction with TLR2 and TLR4.38,41 Through TLR signaling and engagement of different adaptor molecules, biglycan promotes inflammatory, reactive oxygen species (ROS) and sphingolipid pathways to regulate various cytokines and chemokines. 65 Thus, biglycan promotes the production of chemokine (C–X–C motif) ligand (CXCL) 1 and CCL2 via TLR2/4/myeloid differentiation primary response gene 88 (MyD88) to recruit neutrophils and macrophages, whereas the expression of CCL5 via TLR4/TIR-domain-containing adaptor-inducing interferon-β (TRIF) attracts T cells and macrophages. 70 At earlier times, these pathways also involve the activation of sphingosine kinase 1 (SphK1), an enzyme that catalyzes the phosphorylation of sphingosine to sphingosine 1-phosphate. 101 Moreover, biglycan induces the release of mature IL-1β which is modulated in a ROS-dependent fashion.39,102 However, biglycan also plays a limiting role regarding IL-1β production. Biglycan promotes the synthesis and activation of NADPH oxidase (NOX) 2 in macrophages, which inhibits the synthesis of IL-1β. However, the anti-inflammatory effect of biglycan-induced NOX2 activation is counteracted by biglycan signaling through TLR2 and heat shock protein 70 (HSP70) induction. 102
Similar to biglycan, decorin binds directly to both TLR2 and TLR4 receptors, thereby orchestrating pro- and anti-inflammatory cytokines and recruiting macrophages.40,41 Furthermore, decorin suppresses the activity of TGF-β1 and reduces the abundance of oncogenic microRNA-21, thus enhancing the protein level of programmed cell death 4 (PDCD4), which in turn will inhibit the transcription of anti-inflammatory IL-10. 41 Consequently, this proinflammatory state provided by soluble decorin limits the expansion of established tumors. 41 The direct interaction between these two SLRPs and TLRs is thought to be due to the presence within their protein structure of LRR motifs, while the signaling requires the presence of GAG moieties. 38 Interestingly, some PGs may also interact with PAMPs and PRRs. For example, lumican being structurally very similar to decorin and biglycan has up to now not been described as an independent trigger of TLRs but rather as an enhancer of LPS. Therefore, lumican core protein binds LPS and enhances the LPS/TLR4/CD14-dependent proinflammatory response. 103
In addition to intact PGs, proteolytic degradation of two chondroitin sulfate PGs, aggrecan and versican, results in the generation of fragments that are important in cancer progression and are DAMPs. 104 The aggrecan 32-mer fragment, which is generated upon proteolytic degradation of the aggrecan core protein, activates chondrocytes, synovial fibroblasts, and macrophages in a TLR2/MyD88/NF-kB-dependent manner. 37 Activation of TLR2-dependent signaling by the aggrecan 32-mer stimulates the expression of inducible nitric oxide synthases (iNOS), CCL2, IL-1α, IL-6, MMP12, MMP13, and ADAMTS5 in chondrocytes, synovial fibroblasts, and peritoneal macrophages. Similarly, versikine, the 70-kDa N-terminal fragment of versican, activates macrophages resulting in the increase of type I IFN–stimulated genes and macrophage production of IL-6, IL-1β, IL-12, and CCL2. 105 The work of Tang et al. shows that intact versican is recognized by macrophages in a TLR2-dependent manner. 72 When macrophages deficient in TLR2 were exposed to versikine, there was a significant but only partial decrease in IL-6, suggesting that TLR2 along with other PRRs are responsible for macrophage recognition of versikine. 72
Hyaluronic acid (HA), a non-sulfated GAG without a protein core, is capable of regulating the innate immune response via TLRs through different mechanisms which are dependent on its molecular weight.2,63 LMW-HA has been shown to act as an endogenous activator of a TLR2-promoted immune response, whereas high-molecular-weight hyaluronan (HMW-HA) inhibits TLR2 signaling. 42 Moreover, the LMW-HA fragments released upon tissue injury engage TLR2/4/MyD88 and CD44, and activate the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway.42,106,107 Consequently, these signaling pathways potentiate the expression of various cytokines and enzymes that modulate ECM homeostasis (e.g., metalloproteinase [MMP] 12, inducible nitric oxide synthase, and plasminogen activator inhibitor-1). Moreover, HS fragments released by heparanases 108 from perlecan (basement membrane), 109 syndecans, 110 and glypicans 111 (cell surface) act as direct TLR4 agonists.47,62,112 It appears that HS binds also to other TLRs containing the HS-binding motif, such as TLR1, TLR2, or TLR6. More details are addressed in a recently published comprehensive review. 112 On the contrary, HS chains, while still attached within heparan sulfate proteoglycans (HSPGs), modulate the innate immune response by facilitating the LPS-induced CD14/TLR4 activation. 113 In this context, studying the effects of HS biomimetics, synthetic compounds, or regulation of heparanases might bring us closer to the identification of specific drug targets to limit the untoward effects of excessive inflammation.108,114
Other potent activators of TLR4 are the endogenous soluble ECM glycoproteins such as tenascin-C,49,58,115 fibrinogen extravascular deposits, 50 and FN-derived domains51,52,55 (Table 1). FN-derived domains are generated as a consequence of unfolding due to cellular contractility 54 or alternative splicing mechanisms. 53 FN III 13-14, 55 FN extradomain A,51,56,57 the partially unfolded III-1 domain, and FnIII-1c 52 are FN-derived domains revealed to signal through TLRs. For more insights on the structure of FN, please refer to more thematic reviews.116,117
The non-collagenous matrix protein matrilin-2 released during axonal injury has been shown to signal through TLR4 in the central nervous system. 64 Even though the ECM-derived DAMPs share similar TLRs (mainly TLR2 or TLR4), the signaling outcomes differ widely from one DAMP to the other.41,65,118,119
Inflammasomes and ECM-Derived DAMPs Signaling
Another class of PRRs is represented by inflammasomes, which are intracellular innate immune receptors activated by specific ligands to form molecular scaffolds that recruit and activate the cysteine protease caspase-1. 120 Caspase-1 in turn cleaves and matures IL-1β and IL-18 cytokines and is capable of induction of pyroptosis, the inflammatory form of cell death, which is thought to be a mechanism to remove compromised cells.120,121 However, there are a number of molecules that arrest pyroptosis in infected cells to maintain their survival.122,123 Some inflammasomes such as NLR family caspase activation and recruitment domains–containing protein 4 (NLRC4); NACHT, LRR, and PYD domains–containing protein (NLRP) 1; and absent in melanoma 2 (AIM2) are known to be activated by direct interaction with viral or bacterial ligands delivered in the cytosol. 120 NLRP3, NLRP6, and pyrin have been shown to be more sensitive to different cellular biochemical changes or signaling like membrane potential modification, potassium efflux, ROS formation, or metabolic changes.120,121,124–126 Two other human inflammatory caspases, caspase-4 and -5, were shown to be also involved in the innate immune response 127 ; however, their regulation by ECM-derived ligands is not well understood.
At this time, biglycan and LMW-HA are the main ECM-derived DAMPs shown to activate the inflammasome and subsequently caspase-1. 5 To promote the formation of the NLRP3 inflammasome complex, caspase-1 activation, and IL-1β maturation in macrophages, soluble biglycan must first interact with TLR2/4 and the purinergic receptor P2X7 39 (Table 1). However, the exact biochemical mechanism of biglycan-mediated NLRP3 activation is not yet fully clarified. It is conceivable that biglycan alters membrane potential by binding to the ligand-gated ion channel P2X7. 39 Another possible mechanism might be the induction of ROS by biglycan, which is known to be involved in the formation of the inflammasome scaffold. 121 Based on recent study regarding the dual role of biglycan in IL-1β regulation, 102 it appears that while biglycan induces the inflammasome scaffold formation through the NOX1- and NOX4-generated ROS, biglycan might exert anti-inflammatory signaling via NOX2-produced ROS.
Even if soluble biglycan induces the synthesis and the activation of SphK1, thereby enhancing the CCL2 and CCL5 inflammatory cytokines, 101 nothing is known about the biglycan/SphK1 axis in terms of activation of the inflammasome. Nevertheless, studies show that SphK1 activation has an inhibitory effect on inflammasome activation by decreasing the levels of sphingosine, a DAMP that promotes NLRP3-inflammasome-dependent secretion of IL-1β.128–130 However, the hypotheses regarding the direct mechanisms of biglycan-induced inflammasome activation require further studies to be well established.
On the contrary, LMW-HA activates the NLRP3/cryopyrin inflammasome, thereby promoting the maturation of IL-1β through a different mechanism. LMW-HA is internalized by macrophages and subjected to lysosomal digestion into fragments, which in turn activate the inflammasome. 45
HS has not been studied in relation to the inflammasome pathways, even though inflammasomes such as NLRP3, NLRC4, or AIM2 contain HS-binding motifs 112 that should be recognized when HSPGs become engulfed intracellularly. Tenascin-C together with ATP-P2X7R signaling can also activate the inflammasome through TLR4 in epicardium-derived cells following myocardial infarction. 131
HS and RAGE Signaling
RAGE has been initially revealed as the receptor for advanced glycation end products (AGEs) formed after rearrangement of the non-enzymatic glycosylated products.132–134 RAGE is one of the most studied receptors for DAMPs known to recognize ligands like HMGB1, 32 S-100 calcium-binding proteins, 34 myosin fragments, 18 and amyloid-β. 135 Structurally, RAGE possesses an extracellular domain formed by three immunoglobulin domains (V-C1-C2), a single transmembrane domain and cytosolic portions. The V and C1 domains are decisive for the ligand–receptor interaction, whereas intracellular domain is crucial for signal transduction.48,132,136
RAGE expression is augmented during embryonic development; it remains in a basal level in healthy tissues but is reactivated in adults under conditions such as inflammation. 137 Due to natural alternative splicing and cleavage by membrane-associated protease, RAGE has three variants: N-truncated (V-type domain absent), dominant-negative (cytosolic domain absent), and soluble RAGE (transmembrane domain absent). 138 RAGE-dependent signal transduction requires the oligomerization of RAGE on the cell surface, 48 and inhibition of this process reduces mitogen-activated protein kinases (MAPKs) and NF-kB activation. 139 Out of all matrix-derived DAMPs, HS chains bind to RAGE with a receptor: HS stoichiometry of 2:1 and trigger the formation of a hexamer. 48 The binding of HS to RAGE is essential for the receptor activation and downstream Erk1/2 phosphorylation following stimulation with known RAGE ligands like HMGB1, S100A8/A9, S100A12, S100b, or AGE-BSA.48,140,141 Besides HS, RAGE was shown to bind CS chains and promote the pulmonary metastasis of Lewis lung carcinoma and B16 melanoma cells. 142 Based on the potential for RAGE to be involved in chronic inflammation and metastasis, it is considered a therapeutic target. Soluble RAGE is a potential therapeutic tool to limit tissue inflammation and tumor metastasis because it competes with ligands, including AGEs, that bind to RAGE localized on endothelial cells, and inhibits RAGE signaling.33,143–145
Indirect Modulation of Innate Immunity by ECM-Derived DAMPs
TGF-β in Innate Immunity
TGF-β is an extensively characterized cytokine with roles in diverse biological processes such as regeneration, angiogenesis, immune responses, and homeostasis.66,146–148 The origins of TGF-β dysregulation have been of great interest because this has the potential to play a pivotal role in the development of fibrosis. 66 TGF-β pathway activation initially requires release of TGF-β from large latent complexes (LLCs) formed by the latent-associated protein (LAP), TGF-β, and the latent transforming growth factor β binding protein (LTBP). 149 Once released, TGF-β binds to its receptor, which is a tetramer and promotes kinase activity and the downstream Smad-dependent or -independent signaling pathways.66,150 The Smad-independent pathways include the activation of the phosphoinositide 3-kinase (PI3K), Rho GTPases, and various MAPKs.66,150
In addition to its profibrotic effects, TGF-β has been shown to modify the innate and adaptive immune systems with effects on almost every immune cell, including dendritic cells, B cells, NK cells, innate lymphoid cells, and granulocytes. 66 For example, TGF-β promotes the differentiation of macrophages to an M2 or anti-inflammatory phenotype. 151 Thus, TGF-β decreases the NF-kB- and TNF-α production in macrophages by interfering with the TLR2, TLR4, and TLR5 receptors as well as the MyD88 pathway, while TRIF-related adaptor molecule (TRAM)/TRIF signaling remains unaffected. 152 This is mainly due to TGF-β-dependent ubiquitination and proteasomal degradation of MyD88 and the subsequent decrease of its cellular levels. 152 Moreover, the inflammatory anergy of intestinal macrophages is attributable to the action of intestinal stromal TGF-β, which is able to induce IkBα expression, thus inhibiting NF-κB activation in a Smad-dependent manner. 153 However, TGF-β also promotes proinflammatory responses because inhibition of TGF-ß signaling decreases LPS-induced TNF-α production in macrophages. 154
ECM-Derived DAMPs and TGF-β Signaling Crosstalk in Inflammation
Among the ECM-derived DAMPs, decorin is the most extensively studied TGF-β-binding partner, and their interaction results in various biological effects. The proinflammatory response triggered by decorin can be attributed to three different mechanisms such as direct interaction between soluble decorin and the TLR2 and TLR4 receptors, inhibition of the TGF-β signaling, and sequestration of TGF-β by matrix-bound decorin. The direct signaling of soluble decorin through TLR2 and TLR4 is described above and schematized in Fig. 1.
Heparin and HS also bind TGF-β1, suggesting the potential for these GAGs to control the biological activity of this molecule. 155 Heparin and highly sulfated HS were shown to bind TGF-β1 and TGF-β2 but not TGF-β3. 156 The ability of heparin and HS to modulate the biological activity of TGF-β is summarized in a new comprehensive review. 157 However, that ability of GAGs to modulate TGF-β activity in the innate immune response to PAMPs and DAMPs was not addressed in this article.
Highly sulfated hyaluronan molecules have been shown to interact with TGF-β, thereby impairing its bioactivity by blocking the binding of TGF-β1 to its receptors-I and –II and consequently decreasing Smad2 phosphorylation. 158 In addition, the fifth FN type III–like domain of tenascin-C, called TNCIII5, has been shown to bind TGF-β family members with high affinity. 159
ECM-Derived DAMPs Promote an Anti-Inflammatory Response
There is growing evidence that many of the same ECM-derived DAMPs that bind to PRRs and activate a proinflammatory response are also capable of binding to PRRs to promote an anti-inflammatory response.160–162 HA can activate both pro- and anti-inflammatory pathways depending on size with LMW-HA (<250 kDa) promoting the proinflammatory effects of HA, which are described in detail earlier in this review. In contrast to the LMW-HA, studies show that HMW-HA (>1 million Da) interactions with TLRs provide signals of tissue integrity and are important in suppressing the inflammatory response. In one study, HMW-HA interactions with TLR4 in type 2 alveolar epithelial cells (AEC2) promoted an anti-inflammatory response that leads to proliferation of AEC2 cells and repair of lung injury and decreased the pulmonary fibrosis in a mouse model of bleomycin-induced lung injury. 163 In a second study, HMW-HA was found to specifically inhibit the TLR2-dependent signaling of LMW-HA in MH-S cells, an alveolar macrophage cell line. 42
As previously discussed, biglycan promotes a proinflammatory response through activation of TLRs and inflammasomes. 2 In addition to activation of MyD88 signaling pathways after engaging TLR4, biglycan activates the TLR4 co-adaptor molecule, TRIF, activating type I interferon signaling pathways.70,102 Signaling through TRIF is considered to be important in the resolution of inflammation via the expression of type I interferon–stimulated genes. 164 Recent work from the cancer literature shows that intact versican binds TLR2 and differentiates dendritic cells to an anti-inflammatory/immunosuppresive phenotype with increased production of IL-6 and IL-10 and increased expression of IL-6 and IL-10 receptors on the cell surface of these immune cells. 72 The polarization of dendritic cells to an immunosuppressive phenotype—a well-recognized consequence of inflammation associated with cancer—is now considered to be a mechanism contributing to immune evasion by tumor cells.72,165
In contrast to other ECM-DAMPs that activate TLRs causing either a pro- or anti-inflammatory response, proteoglycan-4 (PRG4), also known as lubricin, has been shown to only promote an anti-inflammatory response. Abundant in normal joints, PRG4 decreases in the presence of inflammatory arthropathies, such as osteoarthritis (OA) and rheumatoid arthritis (RA). 166 The binding of PRG4 to CD44, TLR2, and TLR4 on synovial fibroblasts suppresses the production of a number of proinflammatory mediators including cytokines and proteases and decreases the proliferation of synovial fibroblasts when these fibroblasts are treated with TLR agonists or synovial fluid obtained from patients with OA and RA.167–169
Future studies will need to better define the mechanisms whereby ECM-derived DAMPs shape the innate immune response during the resolution of tissue inflammation and the progression from innate to acquired immunity. This will include better characterization of the signaling pathways and transcription factors by which HMW-HA, biglycan, and versican promote an anti-inflammatory response. More research is also needed to better define the role of ECM-DAMPs such as HMW-HA and PRG4 in maintaining tissue homeostasis. Understanding the mechanisms whereby ECM-DAMPs promote an anti-inflammatory response may lead to new treatment strategies to minimize the untoward effects of excessive inflammation.
Over the past two decades, studies have provided mounting evidence of the importance of the ECM in shaping the innate immune response. This is a paradigm shift from the initial concept where the ECM was considered a static network involved in the structural integrity of tissues to a new paradigm where the ECM is dynamic and is able to shape the immune responses to tissue inflammation and injury.170,171 A prime example is the release of ECM components, the so-called DAMPs that bind PRRs and activate select signaling pathways of the innate immune system. The study of the biglycan and decorin has attracted a considerable amount of attention and more importantly resulted in the identification of specific PRRs including TLR2, TLR4, and TLR6, and the NLRP3 inflammasome that are activated resulting in the production of proinflammatory mediators and the acute inflammatory response (Fig. 1).5,40 More recently, studies provide evidence that these same molecules activate PRRs such as TLR2 and TLR4 and play a critical role in promoting an anti-inflammatory response that is critical for resolution of tissue inflammation and the transition to acquired immunity.102,163,165
Despite these advances, there is much that still needs to be addressed. For example, structural modifications of HS and CS side chains of ECM-PGs occur in response to tissue inflammation and injury. However, very little is known about how these modifications alter the ability of ECM-PGs to bind and activate PRRs. It is also interesting that only a limited number of ECM-derived DAMPs have been discovered, raising the question as to whether additional ECM-DAMPs exist. Another potential avenue that needs to be pursued is the use of these molecules as biomarkers that might have diagnostic relevance in certain pathological conditions.40,172–174 Finally, increased knowledge of how the ECM-derived DAMPs trigger the innate response will enable the development of novel therapeutic strategies to limit the deleterious effects of excessive tissue inflammation.
Footnotes
Acknowledgements
We apologize to those researchers whose work could not be cited due to space limitation.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All of the authors contributed to writing the manuscript. CFW, JF, MW, and LS designed the concept of the article (the order and content of the review sections). LS and MVN designed the figure and table. All of the authors provided final approval of the submitted and published versions.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Original research on ECM signaling in the authors’ laboratories was supported by the German Research Council (SFB 815, project A5, SFB 1039, project B02, SFB 1177, project C2, and SCHA 1082/6-1), LOEWE program Ub-Net (all to LS), German Research Council (WY119/1-3), and German Center for Lung Research (to MW).