
Abstract
Idiopathic pulmonary fibrosis (IPF) is a parenchymal lung disease characterized by progressive interstitial fibrosis. The clinical course of IPF can be unpredictable and may be punctuated by acute exacerbations. Although much progress is being made in unraveling the mechanisms underlying IPF, effective therapy for improving survival remains elusive. Longitudinal disease profiling, especially in terms of clinical manifestations in a large cohort of patients, should lead to proper management of the patients and development of new treatments for IPF. Appropriate multidisciplinary assessment in ongoing registries is required to achieve this. This review summarizes the current status of the diagnosis and clinical manifestations of IPF.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a specific type of chronic progressive fibrosing interstitial pneumonia associated with a histopathologic pattern of usual interstitial pneumonia (UIP). Substantial progress in understanding the pathobiology, natural history, and clinical significance of IPF has been made in the past decade, and two new therapeutic options have recently been established. Further understanding of the clinical aspects of the disease is crucial for proper management of patients and the development of novel therapies. This review highlights current research on IPF, especially its diagnosis and clinical manifestations.
Epidemiology and Risk Factors
The reported overall prevalence and incidence of IPF varies from 0.5 to 27.9/100,000 and 0.2 to 8.8/100,000, respectively.1–3 Recently, Hutchinson et al. 4 reviewed previously published population-based studies (1968-2012) on the worldwide incidence of IPF and mortality from IPF. They estimated a conservative incidence range of 3-9 cases per 100,000 per year for Europe and North America. Incidence was lower in East Asia and South America. However, they also pointed out that there are some limitations in directly using these data because of the following reasons.1,5 (1) Only a few of the published reports about the epidemiology of IPF include cases diagnosed using the most recent diagnostic criteria. (2) These data are from different and heterogeneous sources, eg, national registry databases, questionnaire-based studies, and analyses of the registry databases of hospitals and health-care providers. (3) Most of the studies either have major methodological limitations or include only a small cohort of patients. Hence, uniform international diagnostic standards will be crucial in facilitating the collection of more accurate and comparable data in future. Some preliminary reports on several well-organized prospective national registries have already been published,6–10 and these will hopefully yield comparable data on IPF epidemiology from different geographical and cultural areas.
Certain risk factors associated with IPF 11 include cigarette smoking, viral infection, environmental pollutants, chronic aspiration, genetic predisposition, and drugs. However, none of these risk factors adequately explain the extensive remodeling and progressive nature of IPF or the increase in the incidence of fibrosis with advancing age. Studies on families with several affected members (familial pulmonary fibrosis)12–25 have suggested a genetic predisposition to pulmonary fibrosis. Moreover, 2%-20% of the patients with IPF have been reported to have a first-degree relative with interstitial lung disease (ILD).26–28 In a Mexican case–control study, the presence of a family history of pulmonary fibrosis was found to be the most important risk factor for IPF with an odds ratio of 6.1 (95% confidence interval, 2.3-15.9) on multivariate analysis. 28 However, the precise genetic and host susceptibility factors that determine the phenotypic expression and clinical manifestations of sporadic IPF remain unknown. 29
Clinical Presentation
Symptoms and Physical Findings
Dyspnea is the most frequent symptom reported by patients with IPF at the initial visit. Several studies have shown correlations between the severity of dyspnea and quality of life and survival in patients with IPF.30,31 Although a change in the severity of dyspnea has been shown to predict survival, 32 the dyspnea measurement that is most predictive of outcome remains unclear. Cough is also a common symptom in patients with IPF and is more prevalent in patients who have never smoked or in those who have more advanced disease. Cough is considered an independent predictor of disease progression, 33 and severity of cough, which is a common disabling phenotypic component of IPF, is significantly associated with the presence of the minor allele of a MUC5B promoter polymorphism. 34 Clinical features suggestive of an underlying connective tissue disease (CTD), such as arthralgia or sicca symptoms, might also be observed. These signs should be assessed by clinicians including rheumatologists when diagnosing IPF.
Fine crackles, predominantly in the lower posterior lung zones, are commonly reported in patients with IPF, and clubbed fingers are reported in 30%-50% of patients (Fig. 1). The exact cause of clubbing is still unknown, but Kanematsu et al. 35 reported that the presence of clubbing was correlated with the extent of smooth muscle proliferation within areas of fibrotic change in lung biopsy specimens. The majority of patients with IPF are not underweight; however, body mass index has also been associated with survival in these patients. 36

“Clubbed fingers” characterized by hypertrophy and enlargement of the distal phalanges of the hands. The inset shows a slanting view.
Respiratory Physiology
Routine spirometry consistently shows restrictive impairment with reduced lung volumes and decreased diffusing capacity in patients with IPF.32,37-39 However, in a recent Canadian cohort study, one in four patients had normal total lung capacity and more than half of the patients had a normal forced vital capacity (FVC). 40 As the severity of restriction increases, forced expiratory volume in one second/FVC increases and diffusing capacity for carbon monoxide (DLCO) decreases. Longitudinal changes in pulmonary physiology are clearly an important predictor of mortality due to IPF, and several studies have confirmed that declines in lung function, particularly declines in FVC,11,32,39,41,42 total lung capacity, alveolar–arterial gradient, 32 and DLCO,32,39,42 are useful predictors of mortality from IPF. A decline in FVC over a period of 6 or 12 months reliably predicts mortality,11,32,39,42 and a decline in DLCO has been associated with decreased survival, although less consistently.32,43 The results of a 6-minute walk test (6MWT) are weakly correlated with physiological function, and a 24-week decline of greater than 50 m in the 6MWT distance predicts mortality. 44
High-Resolution Computed Tomography Findings
The ATS/ERS/JRS/ALAT 2011 guidelines 11 have assigned a primary diagnostic role to high-resolution computed tomography (HRCT). These guidelines state that HRCT features of IPF should be described as UIP, possible UIP, or inconsistent with UIP. Although a revised guideline has been published in 2015, 45 the HRCT criteria are the same as those outlined in the 2011 version. UIP is characterized on HRCT by the presence of reticular opacities, often associated with traction bronchiectasis.46,47 Honeycombing is common and is critical for making a definite diagnosis. Ground glass opacities are also common, but are usually less extensive than the reticulation. The distribution of UIP on HRCT is characteristically basal and peripheral, but it is often patchy (Fig. 2). The UIP patterns on HRCT are highly accurate for the presence of a UIP pattern on surgical lung biopsy (SLB). However, the presence of coexisting pleural abnormalities, micronodules, air trapping, nonhoneycomb cysts, extensive ground glass opacities, consolidation, or a peribronchovascular-predominant distribution suggests an alternative etiology for the UIP pattern, namely, “inconsistent with UIP pattern.” If honeycombing is absent, and the imaging features otherwise meet the criteria for UIP, the imaging features are regarded as representing “possible UIP” (Fig. 2) and SLB is necessary to make a definitive diagnosis. This HRCT classification based on the 2011 guidelines 11 seems easier to use and is potentially more reproducible than the 2002 classification. 48 In fact, Raghu et al. 49 reported that 79 of 84 patients with possible UIP pattern on HRCT have had a biopsy confirmation of UIP. There is also increasing interest in using HRCT patterns as prognostic determinants. Recently, Romei et al. 50 reported that this HRCT classification based on the 2011 guidelines showed high accuracy in stratifying fibrotic changes because the UIP, possible UIP, and inconsistent with UIP patterns seem to be correlated with different disease progression and mortality rates. However, some potential for poor interobserver agreement must be taken into account. One of the problematic areas identified by several studies51–56 was in differentiating honeycombing from traction bronchiectasis or emphysema. In general, honeycombing can be regarded as being more thick walled, subpleural, and parallel to the chest wall. Emphysema is typically characterized by thinner walls and cystic airspaces that have a propensity to be located further away from the chest wall.52,57
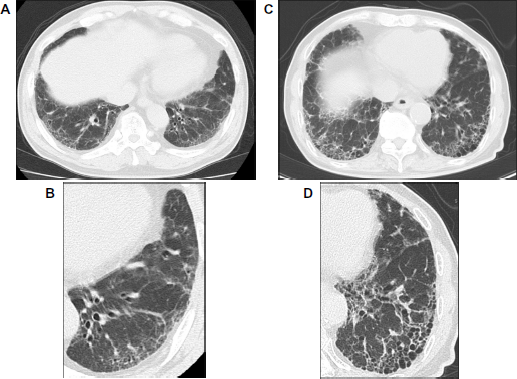
High-resolution computed tomography (HRCT) images demonstrating usual interstitial pneumonia (UIP) pattern and possible UIP pattern. (
Chest radiography is less useful than HRCT in evaluating patients with suspected IPF. However, it is helpful for evaluating disease distribution and serial change in volume loss while following up the patients with IPF.
Biomarkers
Major advances in the understanding of the pathobiology of IPF over the past decade have led to the identification of numerous potentially useful genetic and molecular biomarkers. In addition, there has been increased interest in applying the concept of “precision medicine” to IPF, in particular to search for those genetic and molecular biomarker-based profiles.
On the basis of the type of information they provide, biomarkers are classified into (1) diagnostic biomarkers, (2) disease susceptibility markers, (3) prognostic biomarkers, (4) disease activity markers, and (5) drug efficacy biomarkers. Moreover, IPF biomarkers can be classified on the basis of their association with core mechanistic pathways as follows: (1) epithelial cell dysfunction and senescence, (2) aberrant innate and adaptive immunity, and (3) abnormal lung remodeling. A predisposition of epithelial cells to injury combined with the impaired cellular renewal characteristic of telomere dysfunction may lead to the interstitial changes typical of IPF (SP-A,58,59 SP-D, 58 KL-6, 60 MUC5B, 20 TERT/TERC,21,22 and telomere length 61 ). Aberrations in innate and adaptive immunity may represent important mechanisms in IPF and may define immune-based endotypes (TLR-3, 62 CCL18, 63 anti HSP70, 64 YKL-40, 65 CXCL13, 66 and CD28CD4T cells 67 ). Aberrant matrix remodeling may be an important driver of disease progression in some patients (MMP1, MMP7, 68 periostin, 69 osteopontin, 70 circulating fibrocytes, 71 and markers of MMP activity 72 [a subset of protein fragments generated by extracellular matrix turnover]). A potential role for infections as a cofactor in disease development and progression 73 or as a trigger in disease exacerbation has also been proposed, and a number of other candidate biomarkers have been reported.
Recently, we also reported two potential biomarkers for IPF, namely, plasma CCN2 74 and Mac-2 binding protein. 75 Connective tissue growth factor (CCN2) is a key profibrotic factor associated with transforming growth factor-β. In our cohort, plasma CCN2 levels showed a significantly negative correlation with the six-month change in the FVC of patients with IPF. Meanwhile, Mac-2 binding protein (M2BP) is a cell-adhesive glycoprotein of the extracellular matrix secreted as a ligand of galectin-3 (Mac-2). Recently, a Wisteria floribunda agglutinin-positive-M2BP (WFAt-M2BP) assay developed using a lectin–antibody sandwich immunoassay has shown promise as a new fibrotic marker in liver fibrosis to detect unique fibrosis-related glycoalteration. In patients with IPF, a significant positive correlation was found between serum WFAt-M2BP levels and age; KL-6, neutrophils in bronchoalveolar lavage, reticulation and honeycombing scores obtained using HRCT data; and fibrotic foci scores determined using pathological findings. However, a significant negative correlation was found between serum WFAt-M2BP levels and FVC, %DLCO, and macrophages in bronchoalveolar lavage. Importantly, patients with highserum WFAt-M2BP levels had a significantly worse prognosis than did those with low levels (log-rank test, P = 0.0209). Moreover, a high-serum WFAt-M2BP level was a significant prognostic factor in the Cox proportional hazards regression analysis. Although no universal, validated IPF biomarkers are yet available, the available data regarding the potential use of genetic and molecular biomarkers are promising in predicting prognosis in cases of IPF.
Diagnosis
Diagnosis of IPF depends on the following criteria: (1) exclusion of other known causes of ILD; (2) presence of a UIP pattern on HRCT in a patient who has not undergone SLB; and (3) specific combinations of HRCT and SLB findings in patients who have undergone SLB, as presented in Table 1. Although a precise description of the histopathological criteria behind the guideline is beyond the scope of this review, the designation of definite, probable, or possible IPF based on a combination of HRCT and histology findings is a major advance over the previous statement (Table 2). 48
HRCT criteria for UIP pattern.

Diagnosis of idiopathic pulmonary fibrosis.

A lack of uniform management recommendations for probable and possible IPF, which would be highly prevalent under these new guidelines, could be a problem. 76 However, patients with probable and possible IPF with a UIP pattern tend to have a clinical course that is similar to that of confirmed IPF as defined by the current consensus guidelines. 77 In fact, 94% of patients who met the HRCT criteria for possible UIP also had histologically confirmed UIP. 49 Enrollment of such patients in future trials would greatly increase the number of participants and, therefore, more closely match the trial patients to those in the population likely to be treated if the therapy is found useful.
Recent studies and the international idiopathic interstitial pneumonia (IIP) guidelines of 2013 78 advocate the importance of a multidisciplinary approach to the initial diagnostic assessment of patients with suspected IPF. The members of this discussion include clinicians, radiologists, pathologists, and occasionally, rheumatologists and nurses. Although sometimes difficult to coordinate, this diagnostic approach has been shown to decrease interreader variation in the final diagnosis and increase diagnostic confidence.79,80 Exclusion of other known causes is a difficult, but necessary, step in making a clinical diagnosis of IPF. There are no uniformly validated tools for excluding other known causes. A careful history and physical examination focusing on comorbidities, medication use, environmental exposures, and family history is essential. Evaluating patients thoroughly is particularly important in order to rule out chronic hypersensitivity pneumonitis, which may mimic IPF.81–83 While the clinical history helps, it may be misleading. CTDs can also mimic IPF, both clinically and radiologically.78,84-86 Elimination of specific symptoms and detection of autoantibodies can distinguish CTD from IPF. However, there is growing evidence of substantial overlap, such that patients with IIP may also have signs and symptoms of CTDs including autoantibodies, arthralgias, skin eruptions, or photosensitivity, without fulfilling the criteria for a specific CTD. Remarkably, our recent data have suggested that 10% of patients initially diagnosed with IPF will develop a CTD. 87
For these patients with an “autoimmune flavor,” several disease entities, including undifferentiated CTD,88,89 lung-dominant CTD,90,91 and autoimmune-featured ILD, 92 have been proposed with each provisional criterion. The “European Respiratory Society/American Thoracic Society Task Force on Undifferentiated Forms of Connective Tissue Disease-associated Interstitial Lung Disease” was formed to create consensus regarding the nomenclature and classification criteria for patients with IIP and features of autoimmunity, namely, “interstitial pneumonia with autoimmune features.” 93 The concepts discussed in this statement are intended to provide a platform for the prospective study of such patients.
Staging
There is no widely accepted standardized staging system for IPF. Traditionally, descriptions such as mild, moderate, severe, early, and advanced have been used to stage IPF, and these are usually based on the results of pulmonary function tests. Although this system has been useful in some clinical trials,38,94-97 it is not based on epidemiological or biological data. Recently, it was proposed that the GAP index and staging system (Gender, Age, and two lung Physiology variables, ie, FVC [% of predicted] and DLCO [% of predicted]) were used as a quick and simple screening method for estimating risk in patients with IPF. 98 The GAP index was formulated on the basis of data from retrospective cohort studies at two US centers and one Italian center. A three-step staging system was developed on the basis of this index: stage I, low risk; stage II, intermediate risk; and stage III, high risk. Three-year mortality rate was estimated to be 16.3% in stage I, 42.1% in stage II, and 76.8% in stage III. Although the GAP index has been validated by some studies,99–102 a recent Korean study 103 showed that the GAP model could not accurately predict two- and three-year mortality rates in Korean patients with IPF. This indicates that additional multinational study is needed to validate the applicability and accuracy of this system and to more clearly understand the impact of environmental and genetic differences among affected populations. In Japan, the severity of IPF has been classified into four stages based on the partial pressure of arterial oxygen to guide medical decisionmaking for subsidized care since 1991. This classification system correlates strongly with serial changes in percent vital capacity, DLCO, the incidence of acute exacerbation, and survival. 104 Development of a consensus statement regarding a staging system for enrollment of patients with IPF in random clinical trials should be prioritized.
Natural History of IPF and Acute Exacerbation
Several retrospective longitudinal studies have suggested a median survival time of two to three years from the time of diagnosis of IPF. However, the natural history of IPF is highly variable, and the course of disease in any individual is difficult to predict. The placebo arms of large Phase II and III clinical trials have provided some opportunity to investigate the natural history of lung function decline in patients with IPF,95,97,105-107 but these data are likely biased because the patients enrolled are not a random sample of the general population of patients with IPF. Ongoing nationwide registries may more accurately help elucidate the clinical course and natural history of IPF.6-8,101 Kondoh et al. 108 described acute clinical deterioration in patients with IPF who developed acute influenza-like symptoms, cough, fever, leukocytosis, and progressive hypoxia in the absence of any identifiable infection. Kondoh's criteria for acute exacerbation of IPF (AE-IPF) include progressive dyspnea for one month or less, new pulmonary infiltrates seen on a chest radiograph, worsening hypoxemia, and the absence of an underlying cause, such as infection. In 2007, Collard et al. 109 proposed some modified criteria. The one-year incidence rate of AE-IPF was 8.6%-14.2%.110–113 AE has a highly deleterious impact on the overall survival of patients with IPF. The one-year survival rate from initial diagnosis of AE-IPF has been reported to be 56.2%. 112 A majority of lung biopsy specimens from patients with AE-IPF show acute and organizing diffuse alveolar damage superimposed upon a pattern of UIP, suggesting that similar mechanisms may be involved in the pathogenesis of AE-IPF and fibroproliferative adult respiratory distress syndrome. 114 This histological similarity to fibroproliferative adult respiratory distress syndrome should be noted. Johannson and Collard 115 proposed a new conceptual framework for AE-IPF that de-emphasizes the etiology and emphasizes the presence of diffuse injury as representing clinically significant disease worsening.
Comorbidities
Emphysema
Combined pulmonary fibrosis and emphysema (CPFE) is a distinct clinical phenotype that has different radiological and pulmonary function test results and different prognostic indicators compared to IPF alone.116,117 A significant proportion of patients with CPFE may also present underlying autoimmune disorders. 118 CPFE and IPF may have similar mortality rates, 119 but Sugino et al. 120 reported that CPFE has a much worse prognosis than does IPF alone. CPFE may also be found in patients with lung cancer, and lung cancer may develop in patients with CPFE. 121 Pulmonary hypertension (PH) is also frequently associated with CPFE. 122
Pulmonary Hypertension
The presence of PH with IPF has been linked to poor outcomes in a number of studies.123,124 The rates of prevalence of PH have mainly been reported as a function of the nature of the affected population, 125 and the range is wide (10%-86%); nevertheless, it seems clear that a high prevalence of PH can be expected with severe IPF, especially in patients with CPFE. What remains unclear is whether PH is an adaptive phenomenon or a surrogate for other deleterious aspects of IPF. 126 Some noninvasive methods, including Doppler echocardiography, can provide clues for the diagnosis of PH, but they have limited sensitivity. Although echocardiography can also provide information regarding associated cardiac abnormalities, 127 right heart catheterization remains the gold standard diagnostic test for PH. PH is reportedly amenable to phosphodiesterase-5 inhibitors (sildenafil) therapy, which is known to improve the quality of life and 6MWT distance in patients with right ventricular systolic dysfunction. 128 Dual endothelin receptor antagonists (bosentan) do not show a conclusive effect on mortality or disease progression.97,129 However, previous studies have suggested that patients with PH secondary to IPF might benefit from it more than do patients without PH alone. 45 Ongoing clinical trials may also yield other useful treatments for PH-ILD.
Thromboembolism and Cardiovascular Diseases
Patients with IPF have been reported to have an increased risk of vascular disease. Hubbard et al. 130 reported an increased risk of acute coronary syndrome, angina, and deep-vein thrombosis in the period before the diagnosis of IPF. During the follow-up period, there was a markedly increased risk of acute coronary syndrome and deep-vein thrombosis. Recently, Sprunger et al. 131 reported that the risk of venous thromboembolism in patients who died of pulmonary fibrosis was 34% higher than that in the background population. Those with venous thromboembolism and pulmonary fibrosis died at a younger age than those with pulmonary fibrosis alone, thereby suggesting a link between a profibrotic and a procoagulant state. Nevertheless, clinical trials on the use of warfarin in patients with IPF have yielded conflicting results. 132 Meanwhile, in a Korean cohort, IPF itself was reported to be an independent risk factor for coronary artery disease after adjusting for age, hypertension, diabetes, and hypercholesterolemia, and the prevalence of coronary artery disease in patients with IPF (7%) was two times higher than that in the healthy controls (3%). 133 Dalleywater et al. 134 investigated the risk of cardiovascular diseases using a large UK primary care database. They found that patients with IPF have increased prevalence of cardiovascular risk factors and have an increased risk for ischemic heart disease that cannot be attributed to the increased prevalence of these risk factors alone. However, further research is warranted regarding the biological mechanism behind the increased risk. 135
Gastroesophageal Reflux Disease
The incidence of gastroesophageal reflux disease (GERD) in patients with IPF is higher than that in the general population, and it has been reported to range between 8% and 87%.7,102,136,137 GERD might also play an important role in the development and progression of IPF, including acute exacerbations.138,139 In fact, GERD is a risk factor for microaspiration, which might cause repeated lung injury and worsening of IPF.140,141 Because of these mechanisms, antacid therapy might decrease the risk of acidic microaspiration-associated lung injury or damage. Retrospective, anecdotal data suggest a beneficial role of proton-pump inhibitors in IPF, including the stabilization of lung function and a reduction in the number of episodes of acute exacerbations.136,138,142-144 However, in contrast to the findings of previous studies, the findings of a recent post hoc analysis of randomized controlled trials conducted by Kreuter et al. 145 did not support any beneficial effect of antacid therapy in patients with IPF. A formal test for GERD is needed before the administration of antacid therapy in patients with IPF.
Lung Cancer
The prevalence of lung cancer among patients with IPF has been reported in several studies to range between 3% and 23%, which is relatively lower than the prevalence of other respiratory comorbidities. 146 Nevertheless, the studies that reported mortality and survival among patients with IPF and lung cancer were limited by small sample sizes. Two studies showed no significant difference in median survival between patients with IPF alone and patients with IPF and lung cancer.147,148 Meanwhile, concurrent lung cancer may have a significant adverse impact on survival in patients with IPF. 149 The distribution of lung cancer histologic subtypes (squamous predominant?) and tumor location in patients with lung cancer and IPF might differ from that in the general population, 150 and patients in both the CPFE and IPF groups had a higher risk of lung cancer than did patients in an emphysema group, suggesting that fibrosis is more strongly associated with cancer than emphysema. 121 Finally, from a pathophysiological perspective, IPF and lung cancer have several striking biological similarities, including aberrant cell proliferation as the initiating event, and they share a number of pathogenetic pathways.151–155 The concept of IPF as a neoproliferative disorder of the lung may advance our understanding of the pathogenesis of IPF by helping us approach the disease from the perspective of cancer biology, which is a relatively vast field of knowledge.
Conclusions
In this review, we have discussed the diagnosis and clinical manifestations of IPF. IPF is a progressive and ultimately fatal disease, but its course in individual patients is extremely variable. Advances in the understanding of IPF via longitudinal disease phenotyping in a large cohort of patients, especially with the implementation of current nationwide registries, should afford an opportunity to overcome the remaining barriers to elucidating the pathophysiology of IPF and development of new treatments.
Author Contributions
Conceived the concepts: YN. Analyzed the data: YN. Wrote the first draft of the manuscript: YN. Contributed to the writing of the manuscript: YN, TS. Agree with manuscript results and conclusions: YN, TS. Jointly developed the structure and arguments for the paper: YN, TS. Made critical revisions and approved final version: YN, TS. Both authors reviewed and approved of the final manuscript.