
Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive lung disease with a prognosis that can be worse than for many cancers. The initial stages of the condition were thought to mainly involve chronic inflammation; therefore, corticosteroids and other drugs that have anti-inflammatory and immunosuppressive actions were used. However, recently, agents targeting persistent fibrosis resulting from aberrant repair of alveolar epithelial injury have been in the spotlight. There has also been an increase in the number of available antifibrotic treatment options, starting with pirfenidone and nintedanib. These drugs prevent deterioration but do not improve IPF. Therefore, nonpharmacologic approaches such as long-term oxygen therapy, pulmonary rehabilitation, and lung transplantation must be considered as additional treatment modalities.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, occurring primarily in adults and is limited to the lungs. It has been associated with a histopathologic and/or radiologic pattern of usual interstitial pneumonia. 1 The prognosis is very poor, with a mean survival of about 2.5-5 years after definite diagnosis - a harsh prognosis that makes it inappropriate to refer to IPF as a benign disease. 2 The natural history of IPF is highly variable and the course of the disease for each individual patient is difficult to predict. Some patients experience rapid decline, others progress much more slowly, and some have periods of relative stability interspersed with acute deteriorations (Fig. 1). Once an acute exacerbation occurs, recovery is extremely difficult. Furthermore, because the condition essentially involves structural changes and develops in elderly people, treatment of complications is very challenging for a variety of reasons, including cardiovascular events, pulmonary hypertension, lung cancer, and so on. In recent years, the introduction of pirfenidone and nintedanib treatment has led to many attempts to develop similar drugs for IPF. Although many drugs for IPF have been validated in clinical trials, no therapeutic methods have led to cure. In this article, we aimed to discuss the present treatment approaches and prognosis of IPF, with focus on the available drug options and nonpharmacologic approaches.

Variable clinical course of IPF.
Guidelines on IPF Diagnosis and Treatment before 2015
Policies for diagnosis and management of IPF were accepted based on the international consensus statement by the American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Asociación Latinoamericana de Tórax (ALAT) in 2011. 1 In this guideline, the period of literature search was from 1996 to 2010, when there was almost no evidence on IPF treatment. Because there were no therapeutic methods providing significant short-term effects and because the disease is fundamentally a chronic progressive condition, the main objective has been prevention of disease progression over time. Therefore, IPF therapies must include ways to not only improve symptoms but also ensure adequate clinical stability.
Initially, corticosteroids and immunosuppressants were used to treat IPF because chronic inflammation was believed to be the cause of persistent fibrosis in the early stages of the condition. However, opinion has gradually changed to that of abnormal repair of alveolar epithelial injury leading to persistent fibrosis, 3 which should be the principal concern of disease management. 4 Therefore, pirfenidone and other antifibrotic agents have taken center stage; since 2004, large-scale clinical studies on these drugs have been conducted. Except for pirfenidone and nintedanib, most of the recently evaluated drugs such as N-acetylcysteine (NAC) were shown to be not efficacious (Table 1). In accordance with this change in the concept of pathophysiology, guidelines on treatment have been updated in 2015. 5
Overview of major clinical trials undertaken in IPF.
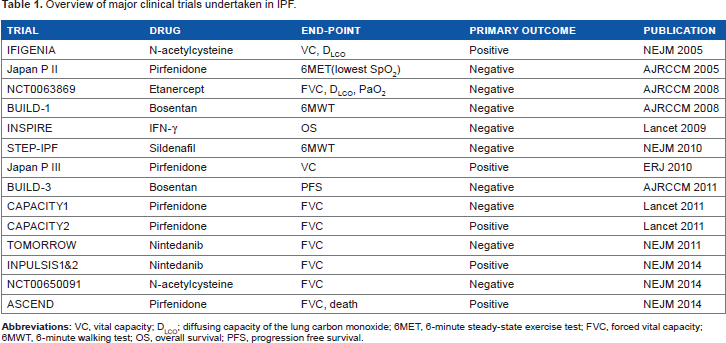
Pharmacologic Therapies
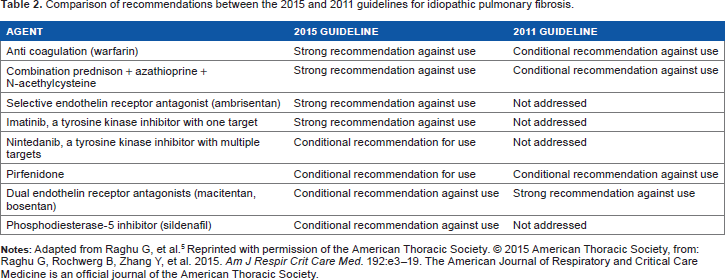
In the ATS/ERS/JRS/ALAT statement in 2011, there were no pharmacologic therapies shown to have clear and satisfactory results. Nevertheless, the 2015 updated guideline described conditional recommendations, based on some clinical studies, about the benefits and disadvantages of certain drugs for IPF (Table 2). However, even if a certain drug was recommended by some, the question of individuals who should be considered for pharmacotherapy remains uncertain. The patients included in the trials had mild to moderate disease. On the other hand, severe patients should be maintained on pharmacotherapy and should be considered to receive nonpharmacologic therapies.
Comparison of recommendations between the 2015 and 2011 guidelines for idiopathic pulmonary fibrosis.
Reduction of mortality is a desirable end point to assess the efficacy of these drugs, but this would be costly and entail analyzing a large number of cases. For this reason, respiratory function and exercise tolerance tests have been adopted as surrogate end points. Forced vital capacity (FVC) has often been used as the main end point in recent clinical studies. Low baseline values, as well as 6- and 12-month decreases in FVC, were reported to correlate with mortality rates. 6 FVC change of 10% or more was a definite prognostic factor in IPF. 7 Moreover, although IPF prognosis is determined by various factors, FVC is reproducible and might be considered the most suitable surrogate end point for vital prognosis. 8 In our opinion, standardizing differences, including race, is one of the challenges when planning international clinical studies.
Pirfenidone
Pirfenidone is a low-molecular-weight compound with anti-inflammatory effects, and it has been found to have antifibrotic action as well. The antifibrotic property of pirfenidone is believed to depend on its ability to inhibit fibroblast proliferation, collagen biosynthesis, and production of transforming growth factor beta and tumor necrosis factor alpha and on its ability to induce anti-inflammatory and antioxidant effects.9–12 A placebo-controlled phase II randomized controlled trial (RCT) was conducted. 13 The total number of cases was 107, and they were categorized into a group (n = 72) receiving pirfenidone (1800 mg/day) and a placebo group (n = 35). There was no difference between the two groups in terms of the main end point, which was change in lowest oxygen saturation from baseline after a 6-minute exercise test (6-minute walk test or 6-MWT); however, vital capacity (VC) significantly improved in the pirfenidone group. 13 Considering these results, a placebo-controlled phase III RCT was conducted. 14 Three groups according to pirfenidone doses were assigned: high dose (1800 mg/day) (n = 108), low dose (1200 mg/day) (n = 55), and placebo (n = 104). At 52 weeks, significant differences were observed in the VC decline from baseline between the placebo (-160 mL) and high-dose (-90 mL) groups, as well as between the low-dose (-80 mL) and the placebo (-160 mL) groups. Based on these results, pirfenidone was approved in October 2008 for IPF therapy in Japan; this was the first approval for IPF drug marketing in the world.
On the other hand, the CAPACITY (studies 004 and 006) clinical trials 15 on pirfenidone for IPF were conducted in parallel at multinational institutions. Both studies were placebo-controlled, phase II RCTs. In the CAPACITY study 004, pirfenidone was shown to be effective based on a significant difference in the main end point, which was change in %FVC from baseline to 72 weeks (pirfenidone group, -8.0% versus placebo group, -12.4%). In contrast, study 006 did not show a similar significant change in baseline %FVC at 72 weeks (pirfenidone group, -9.0% versus placebo group, -9.6%). Based on these results, the US Food & Drug Administration (FDA) withheld the approval of pirfenidone in May 2010, and further clinical study was required. On the other hand, in Europe, the drug began to be marketed in Germany in September 2011; afterward, the number of countries that approved pirfenidone use gradually increased.
A phase III study (ASCEND) 16 was conducted in North America and Oceania to verify the effects of pirfenidone on 555 patients with IPF. After 52 weeks, baseline indexes of IPF progression, including FVC, 6-minute walk distance, and progression-free survival, improved in the pirfenidone group compared with the placebo group (change in FVC: pirfenidone group, -235 mL versus placebo group, -428 mL). In this study, the authors analyzed mortality in the pooled population from the ASCEND 16 and CAPACITY 15 trials and showed that pirfenidone reduced the risk of death at 1 year by 48%, compared with placebo. Considering the acceptable results of the ASCEND study, 16 pirfenidone was approved for marketing by the FDA in October 2014.
The commonly reported adverse events of pirfenidone were gastrointestinal (nausea, dyspepsia, vomiting, and anorexia), skin-related (rash and photosensitivity), and neurologic (headache and dizziness) symptoms; fatigue; and elevated hepatic enzyme levels. These events were generally mild or moderate in severity, in addition to being dose dependent to some extent. Based on our experience, we initially prescribe pirfenidone at 600 mg/day before gradually (at least every 2 weeks) increasing to the standard dose of 1800 mg/day. In case of significant adverse events, we reduce the dose to 1200 mg/day.
Nintedanib
Nintedanib blocks the signal transduction pathway of vascular endothelial growth factor receptors, platelet-derived growth factor receptors, and fibroblast growth factor receptors (triple kinase inhibitor)17,18 to inhibit progression of IPF and worsening of respiratory function. In fact, in a phase II study (TOMORROW), 19 nintedanib was found to inhibit the decline in pulmonary function and acute exacerbation in patients with IPF (change in baseline FVC at 52 weeks: nintedanib group, -60 mL versus placebo group, -190 mL). Furthermore, two randomized, double-blind, phase III reproducibility studies (INPULSIS-1 and -2) were conducted for more than 52 weeks to evaluate the efficacy and safety of twice-daily administration of nintedanib (150 mg) in patients with IPF. 20 The main end point was annual rate of decline in FVC, and the secondary end points were time to first acute exacerbation and changes in the St George's Respiratory Questionnaire (SGRQ) score. A total of 1066 cases were randomly assigned with a nintedanib:placebo ratio of 3:2. The annual rate of FVC decline from baseline was significantly suppressed after 52 weeks in both INPULSIS-1 (nintedanib group, -114.7 mL versus placebo group, -239.9 mL) and INPULSIS-2 (nintedanib group, -113.6 mL versus placebo group, -207.3 mL). Differences between the nintedanib and placebo groups in terms of time to first acute exacerbation and SGRQ score were not significant in INPULSIS-1 but were significant in INPULSIS-2. Because the results of these studies were considered acceptable, the FDA approved nintedanib for IPF treatment on the same date that pirfenidone was approved (October 2014). To date, many of potentially effective options are in the development stage for the treatment of IPF.
The most frequent adverse event of nintedanib is diarrhea. In the CAPACITY trial, 15 more than 90% of patients experienced diarrhea, but only less than 5% were compelled to discontinue nintedanib. Another common adverse event is elevated hepatic enzyme levels. The standard dose of nintedanib is 150 mg twice a day, but in case of significant adverse events, the dose may be reduced to 100 mg twice a day.
Corticosteroids and Immunomodulators
A combination therapy of prednisone, azathioprine, and NAC is widely used for IPF; however, the safety and effects of this regimen have not been well understood until recently. Therefore, a randomized, double-blind, placebo-controlled PANTHER-IPF study was conducted by the US National Institutes of Health on 35- to 85-year-old patients with mild-to-moderate IPF who were assigned to the following groups: prednisone + azathioprine + NAC; NAC only; and placebo. 21 The primary outcome was change in baseline FVC after 60 weeks. In the interim analysis, the number of deaths was eight in the combination group and one in the placebo group. The risk of death was significantly high, and there was a need for frequent and unscheduled hospital admissions (23 cases versus seven cases, respectively). Based on the results of interim analysis, the Data and Safety Monitoring Board recommended discontinuation of the three-drug combination use. In addition, the updated treatment guidelines for IPF strongly recommended against use of corticosteroids and immunosuppressants with NAC for IPF. 5
N-Acetylcysteine
Oxidative stress causes alveolar epithelial cell injury, which leads to lung fibrosis. In normal individuals, redox balance is maintained by superoxide dismutase and glutathione; this balance is disturbed in patients with IPF. NAC not only has antioxidant properties as a glutathione precursor, but it can also act as a direct reactive oxygen scavenger, which is thought to mediate its antifibrotic effect. In the IFIGENIA study conducted in Europe, comparison was made between an NAC group (prednisone + azathioprine + NAC) and a control group (prednisone + azathioprine). 22 In this study, reduction of the main end points, VC and diffusing capacity of the lungs for carbon monoxide (DLCO), was significantly inhibited in the NAC group. On the other hand, there was minimal myelosuppression in the NAC group because glutathione cell concentration was increased by NAC. In addition, liver cell injury was observed, and the side effects commonly associated with azathioprine administration were reduced. On the other hand, comparative evaluation between placebo and NAC alone in the PANTHER-IPF study disclosed no significant differences in the following: rate of FVC change from baseline to 60 weeks of treatment, survival, and frequency of acute exacerbation. 22 Therefore, administration of NAC alone has no significant benefit in IPF. However, in Japan, therapy with combined pirfenidone and inhaled NAC remains a possibility in the clinical setting.
Antacid Therapy
Gastroesophageal reflux disease, including asymptomatic cases, has been observed in more than 90% of patients with IPF. 23 Abnormal reflux is a risk factor for aspiration, which could cause pneumonia and contribute to chronic airway inflammation and fibrosis. In addition, aspiration pneumonia can worsen IPF and could be a risk factor for acute exacerbation. Therapy with antacids, such as proton pump inhibitors (PPIs) and histamine-2 receptor antagonists (H2RAs), may decrease this risk for aspiration pneumonia-associated lung injury or damage. A retrospective analysis of longitudinal cohorts suggested benefit for IPF patients who received PPIs and H2RAs (hazard ratio [HR]: 0.47; 95% confidence interval [CI]: 0.24-0.93; adjusted analysis). 24 Another aggregate analysis of data from three RCTs showed a significantly smaller decrease in FVC in IPF patients who received antiacid treatment at baseline. 25
Comparison between Pirfenidone and Nintedanib
As described earlier, the FDA approved both pirfenidone and nintedanib for the treatment of IPF. Evidence for changes in FVC showed a significantly slower decline in patients treated with pirfenidone or nintedanib compared with placebo. At present, there is no clear evidence regarding which drug is superior. A recent systematic review that indirectly compared both drugs based on network meta-analysis reported slower decline in FVC in patients treated with nintedanib compared with those who received pirfenidone. 26 Nintedanib was significantly better than placebo in decreasing acute exacerbations, whereas pirfenidone was significantly better than placebo in decreasing mortality. By indirect comparison, all-cause mortality was lower with pirfenidone than with nintedanib, but this was not significant (odds ratio [OR]: 1.39; 95% CI: 0.70-2.82). Based on these results on clinical efficacy, the superior first-line therapy for IPF remains to be determined. On the other hand, profiles of side effects are slightly different between the two drugs. Gastrointestinal and skin-related events were more common in the pirfenidone group, 16 whereas diarrhea and liver dysfunction were more common in the nintedanib group. 20 Taking into account these various factors, the choice between pirfenidone and nintedanib for IPF must be individualized.
Nonpharmacologic Therapies
Long-Term Oxygen Therapy
Pronounced hypoxia during physical exertion is a characteristic finding in IPF patients. In fact, hypoxia during the 6-MWT is related to prognosis. 27 There are no data on the use of long-term oxygen therapy in patients with IPF, but it was strongly recommended in the international guidelines by the ATS/ERS/JRS/ALAT. 1
Lung Transplantation
The first successful lung transplantation was conducted in 1983 on a 58-year-old man with IPF. 28 Because IPF has a poor prognosis and no supereffective drugs exist, lung transplantation is commonly considered for patients with moderate to severe disease. 1 However, it is very difficult to determine when a patient can be registered in the transplant list. In 2006, the International Society for Heart and Lung Transplantation released guidelines on the selection of lung transplant candidates, as follows: predicted DLCO of less than 39%; 10% or greater decrease in FVC during 6 months of follow-up; a decrease in oxygen saturation below 88% during 6-MWT; and honeycombing on high-resolution computed tomography scan (fibrosis score of >2). 29 Age more than 65 years is a relative contraindication. In patients with underlying IPF, superiority between bilateral and single lung transplantation is unclear. 5 The 5-year survival rate after lung transplantation in IPF was estimated at about 50%. 30
Pulmonary Rehabilitation
Pulmonary rehabilitation including aerobic conditioning, strength and flexibility training, educational lectures, nutrition advice, and psychosocial support may be beneficial for IPF patients, especially to improve activities of daily living (ADL) and quality of life (QOL). 31 The effects of pulmonary rehabilitation may be more effective in patients with worse baseline functional status. 32 Pulmonary rehabilitation is weakly recommended in the international guidelines of the ATS/ERS/JRS/ALAT because the long-term benefit of pulmonary rehabilitation remains unclear and may not be reasonable in a minority.
Expected Novel Therapeutics
Many molecules are currently in different stages of development for use in the treatment of IPF. Table 3 presents an overview of the most advanced molecules and pathways being targeted. 33
Recent clinical trials on idiopathic pulmonary fibrosis.

The diversity of the pathways being targeted these days, when compared with previous studies, reflects our improved understanding of disease pathobiology. Especially, therapies with monoclonal antibodies in addition to various inhibitors are increasing. Based on the different mechanisms of action between nintedanib and pirfenidone, it would be worthy to evaluate the combined efficacy of both molecules. 34 A recent randomized, double-blind, phase II, dose-escalation trial on nintedanib plus pirfenidone 35 showed that pirfenidone tended to decrease the maximum plasma concentration of nintedanib; on the other hand, nintedanib had no effect on the pharmacokinetics of pirfenidone. Further studies are needed to evaluate the efficacy and safety of this combined therapy. Results of a clinical trial (NCT01872689) on a combination therapy with pirfenidone and lebrikizumab (anti-IL-13) are likewise anticipated.
Prognosis
Table 4 shows the prognostic factors in IPF patients. 1 The definite indexes of IPF prognosis are factors that change over time; these include symptoms, respiratory function, and imaging. 1 These are important points to consider when examining the clinical progression of IPF.
Prognostic factors in idiopathic pulmonary fibrosis patients. (Adapted from Raghu G, et al. 1 )
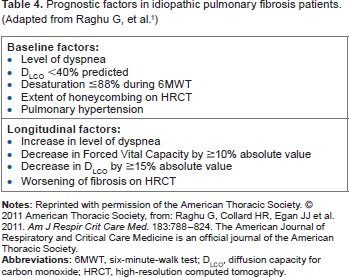
The cause of IPF is still unknown and it is thought to be a heterogeneous condition. Even with a slow start, the disease can sometimes progress rapidly. 36 In addition to having variable progression, IPF remains an incurable disease with poor prognosis. Studies are currently under way to identify the heterogeneous conditions of the disease. Additional and new end points are being anticipated in the future. Currently, the end point used to judge treatment efficacy is clinical improvement and stability of disease. Several studies are being undertaken to develop novel markers of poor prognosis. However, as shown in Table 2, many large-scale studies on several drugs have shown negative results. Unfortunately, as long as there is no breakthrough in drug development, there will be no chance to improve prognosis in IPF.
In general, the reported results on prognosis were based on studies in which the median duration of follow-up was 2.5-5 years. 2 Further, the prognosis of IPF was even poorer than that of many types of cancer. 37 This mortality outcome in IPF has been due to acute exacerbation in many of the cases. In Japan, acute exacerbation has become the most frequent cause of death in up to 40% of cases. 38 However, in some instances, the disease may unpredictably slow down after a rapid progression (Fig. 1). At present, prediction of individual clinical course is not possible; therefore, efforts must be made to find simple and accurate prognostic markers. One easy, simple, and useful marker that has been proposed for predicting IPF prognosis is the GAP score risk prediction model, which includes four factors: gender (G), age (A), and two lung physiologic (P) variables (FVC and DLCO). 39 Since G and A are nonmodifiable variables, improvement of pulmonary function by novel pharmacologic and nonpharmacologic therapies must be developed to improve the prognosis of IPF.
Conclusions
IPF is an intractable respiratory disease with poor prognosis. Several novel therapeutic drugs have been developed, but only a few have demonstrated efficacy in RCTs. These promising drugs include pirfenidone and nintedanib, which are presently available in many countries. Several other drugs are presently undergoing either clinical studies or basic research in the nonclinical stages. In the near future, we expect breakthrough therapies that can lead to improvement of survival in IPF patients.
Author Contributions
Conceived and designed the review article and jointly developed the structure and arguments for the paper: HF, TK, AA. Wrote the first draft of the manuscript, with contributions from AA: TK. All authors agreed with results and conclusions, made critical revisions, and approved the final version of the manuscript.