
Abstract
The use of molecular biomarkers assures that breast cancer (BC) patients receive optimal treatment. Established biomarkers, such as estrogen receptor, progesterone receptor, HER2, and Ki67, have been playing significant roles in the subcategorization of BC to predict the prognosis and decide the specific therapy to each patient. Antihormonal therapy using 4-hydroxytamoxifen or aromatase inhibitors have been employed in patients whose tumor cells express hormone receptors, while monoclonal antibody to HER2 has been administered to HER2-positive BCs. Although new therapeutic agents have been developed in the past few decades, many patients still die of the disease due to relapse; thus, novel molecular markers that predict therapeutic failure and those that can be targets for specific therapy are expected. We have chosen four of such molecules by reviewing recent publications, which are cyclin E, B-Myb, Twist, and DMP1β. The oncogenicity of these molecules has been demonstrated in vivo and/or in vitro through studies using transgenic mice or siRNAs, and their expressions have been shown to be associated with shortened overall or disease-free survival of BC patients. The former three molecules have been shown to accelerate epithelial-mesenchymal transition that is often associated with cancer stem cell-ness and metastasis; all these four can be novel therapeutic targets as well. Thus, large prospective studies employing immunohistochemistry will be needed to establish the predictive values of these molecules in patients with BC.
Keywords
Introduction
Breast cancer (BC) is the leading cause of cancer-related deaths in the United States with more than 230,000 new diagnoses expected in 2014 and almost 40,000 deaths. 1 Although molecular markers, such as hormone receptors (estrogen receptor [ER]/progesterone receptor [PR]), HER2, Ki67, DNA ploidy, and %S phase, have been used to classify the heterogeneous disease into five categories to predict the prognosis and determine the treatment modalities,2–5 current diagnoses and therapies are incomplete because numerous patients die of relapsed disease; thus, improved diagnosis using novel molecular markers of stem cell evaluation to decide therapeutic strategy, gene expression, and microRNA (miRNA) profiling is expected.6–8 A number of research studies have been done to identify novel biomarkers from cell cycle regulators, onco-genes, and tumor suppressor genes that are critically involved in carcinogenesis to improve diagnosis and treatment for BC.
Progression through the cell cycle is driven by cyclin-dependent kinases (CDKs) whose catalytic activity and substrate specificity depend on their association with regulatory subunits called cyclins. D-type cyclins (cyclins D1, D2, and D3) are the first cyclins that are induced in response to mitogens. They bind and activate Cdk4/6 to phosphorylate the retinoblastoma (RB) family proteins, regulating the G1/S-phase transition.9–11 The cyclin D-CDK4/6 complexes also titrate the CDK inhibitors p21Cip1 and p27Kip1 and activate cyclin E/Cdk2, independent of the catalytic activities of CDKs.12–14 The cyclin E/CDK2 complex completes the phosphorylation of pRB and histone deacetylases (HDACs), relieving E2F/DPs from their negative constraint. p27Kip1 is phosphorylated by cyclin E/CDK2 for proteasomal degradation, 15 and when the levels of p27Kip1 decrease to a certain threshold, the G1-S progression becomes irreversible (called restriction point) and cells enter the S phase of the cell cycle.15,16
The E2F family transcription factors (TFs) play essential roles in cell cycle progression and DNA replication.17–19 They can be divided into two major subgroups based on their function and mechanism of action. E2Fs1–3a, the activating E2Fs, are required for the transactivation of target genes involved in the G1-S phase transition and, hence, for accurate progression through the cell cycle. 17 In contrast, E2F3b and 4–8 possess repressive activity, and their major roles have been considered to be the induction of cell cycle exit and differentiation rather than cell cycle progression.17–19 The target genes for E2Fs include genes that are essential for G1-S progression: cyclins E/A, DHFR, TK, TS, POL, and CDC2 and TFs B-Myb, c-Myb, Dmp1, E2F1, and E2F3a.16–20
Cyclin E plays a critical role in G1/S transition by phosphorylating Rb and facilitating E2F:DP release. Cyclin E/Cdk2 phosphorylates p27Kip1 to accelerate its proteasomal degradation, which is essential for mammalian cells to enter the S phase of the cell cycle.13–16 Its expression is associated with not only accelerated cell proliferation but also histone mRNA synthesis/chromatin remodeling through phosphorylation of NPAT/p200 and centrosome amplification through phosphorylation of nucleophosmin and CP110. These chains of events contribute to the malignant phenotypes of tumor cells, which are linked to the induction of chromosomal instability. 21 Cyclin E1/E2 amplifications are key oncogenic events in numerous cancers, especially those arising in the ovary (12–21%), esophagus (14%), and stomach (15%).22–26 The link between cyclin E and poor prognosis is well established in breast and lung cancers but is likely to be observed in other cancers as well.22,27,28 Ectopic expression of cyclin E bypasses the need for CDK4 or CDK6 activity to initiate the S phase,29,30 and it is therefore assumed that amplification of E-type cyclins will bypass the physiological requirement for CDK4/6 activity to initiate the expression of E-type cyclins and thus oncogenic cyclins. Deregulation of CDK2, the catalytic partner for cyclin E, occurs frequently in human cancers; 31 hence, selective inhibition of proteins regulating cyclin/CDK complexes is a strategy against cancer.32,33
The use of two different promoters and different reading frames at the CDKN2 locus provides the generation of two independent transcripts, namely, INK4a and ARF, with tumor suppressor activity. 34 p19Arf (p14ARF) directly binds to Mdm2 (HDM2), sequesters Mdm2 to the nucleus and neutralizes its activity, and thereby activates p53.34–36 The Arf induction by potentially harmful growth-promoting signals forces early-stage cancer cells to undergo p53-dependent and p53-independent cell cycle arrest, apoptosis, or autophagy, thus providing a powerful mode of tumor suppression.34–36 As the INK4a/ARF locus regulates both RB and p53 pathways in human cancer, it is one of the most frequently disrupted loci in human cancers, second only to the p53 locus. The mechanism of gene inactivation involves gene mutation, promoter methylation, gene deletion, aberrant splicing, and others.35,36 This locus is also inactivated by numerous transcriptional repressors, such as Bmi1, Twist1, Ezh2, Tbx2/3, Pokemon, and Geminin (Fig. 1; ref. 37), which play essential roles on epithelial-mesenchymal transition (EMT), stem cell-ness, and metastatic ability of cancer cells.
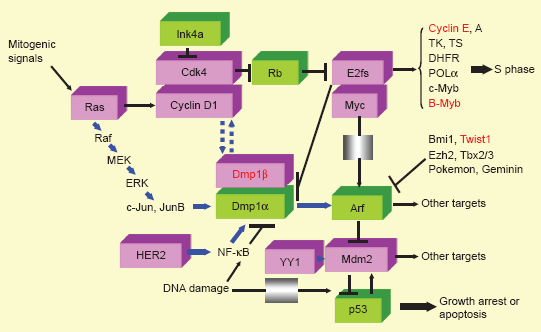
Signaling pathways involving the molecules reviewed. Molecules in pink boxes have oncogenic activities, while those in green boxes have tumor suppressive functions. Molecular markers that are reviewed in this article are shown in red to demonstrate where they work in oncogenic signaling. Mitogenic signals mediated by Ras induce Fos/Jun as early growth response genes, which, in turn, transactivate the cyclin D1 promoter and increase the protein. Cyclin D1 binds to Cdk4 and phosphorylates Rb and HDACs, releasing E2F/DPs from their negative constraint, and the cells enter S phase of the cell cycle.11–16 Cyclin E and B-Myb are both direct targets of E2F/DPs and are explained in this review. Dmp1 is also a target for E2F/DPs. 20 There are two classes of naturally occurring Cdk inhibitors: the Ink4 family proteins (p15, p16, p18, and p19) directly bind and antagonize the activities of Cdk4/6, while Cip/Kip family proteins (p21, p27, and p57) are pan-Cdk inhibitors for cyclin D/Cdk4/6, cyclin E/Cdk2, cyclin A/Cdk2 (or Cdc2), and cyclin B/Cdc2.13–16 The Arf/Ink4a locus generates two independent tumor suppressor genes p19Arf and p16Ink4a that regulate the p53 and Rb pathways, respectively.34–36 Arf is induced by potentially oncogenic signals stemming from the overexpression of oncogenes, such as c-Myc, E2F1, and activated Ras, which quenches inappropriate mitogenic signaling by diverting incipient cancer cells to undergo p53-dependent growth arrest or cell death.34–36 In total, 30–50% of human BCs overexpress INK4a/ARF repressors (eg, Bmi1, Twist1, Ezh2, Tbx2/3, Pokemon, and Geminin)37,181,182 to inactivate the tumor suppressive locus. Dmp1α directly binds and activates the Arf promoter and induces cell cycle arrest in an Arf-dependent fashion in the mesenchymal cells.40,183–188 Dmp1α also physically interacts with p53 and neutralizes all the activities of Mdm2 to activate the p53 pathway. 41 Both Dmp1-/- and Dmp1 +/- mice show hypersensitivity to develop tumors in response to carcinogen and γ-irradiation.51,52 D-type cyclins inhibit Dmp1α's transcriptional activity in a Cdk-independent fashion when E2Fs do not bind to the same promoter; 120 however, cyclin D1 cooperates with Dmp1α to activate the Ink4a and Arf promoters to eliminate incipient tumor cells.44,185 YY1 binds to Mdm2 to accelerate Mdm2-mediated polyubiquitination of p53. 189 The Dmp1 promoter is activated by the oncogenic Ras-Raf-MEK-ERK-Jun 44 or HER2-PI3K-Akt-NF-κB 45 pathways, and thus, Ras- or HER2-driven carcinogenesis is accelerated in Dmp1-null mice. The DNA damage represses the Dmp1 promoter through activation of NF-κB suggesting dual regulation of the Arf-p53 pathway by NF-κB via different signaling cascades. 46 Dmp1α induces both Ink4a and Arf proteins in vivo, and thus, Dmp1α-transgenic mice can be a novel model of aging. 175 DMP1β is an oncogenic splice variant from the DMP1 locus;59,173 the DMP1β/α ratio is significantly elevated in human BC and is associated with poor prognosis of patients. 59 Both cyclin D1 and Ki67 proteins are upregulated in tumors from MMTV-DMP1β V5His mice, suggesting that they are critical targets. 59 Cyclin E, B-Myb, Twist, and DMP1β are frequently overexpressed in human cancers (breast [this review], lung,190–195 and others) and are associated with aggressive disease/shorter DFS, and thus are novel targets for molecular therapy.
The Myb-like TF DMP1 (cyclin D-binding Myb-like protein 1; DMTF1) governs the activity of the ARF-p53 pathway by binding to the ARF promoter and through physical interaction with p53 (see Refs. 37–39 for reviews). The hDMP1-ARF-MDM2-p53 pathway provides cell autonomous tumor surveillance that detects and forces early-stage cancer cells to undergo senescence and/or apoptosis to prevent the development of cancer.40–43 The Dmp1 gene is a direct target of E2Fs and is transcriptionally repressed through direct binding of E2Fs to the 5′ leader sequence during S to G2/M phase of the cell cycle. 20 Mitogenic signals from oncogenic Ras 44 and HER2/neu 45 have been shown to activate the Dmp1 promoter, while physiological mitogens 20 as well as genotoxic stimuli mediated by NF-κB 46 cause its transcriptional repression. Overexpression of the Wilms tumor gene (WT1) has been reported in human leukemic cells, regardless of the disease subtype, and thus can be used for the detection of minimal residual disease.47–49 The human DMP1 gene (hDMP1) expression is suppressed by WT1 in leukemic cells via direct binding to an EGR/SP1 site, delineating a new oncogenic WT1 mechanism of control in the hematopoietic system. 50 Eμ-Myc-, K-ras LA_ , and HER2/neu-driven tumor development was significantly accelerated in both Dmp1 +/- and Dmp1 -/- mice, with no significant differences in the survival between the two cohorts, suggesting that Dmp1 is a haplo-insufficient tumor suppressor.45,51–53 In Eμ-Myc lymphomas, the combined frequencies of p53 mutation and Arf deletion in mice of Dmp1 +/- or Dmp1 -/- background were significantly lower than that in Dmp1 +/+ littermates, indicating that Dmp1 is a physiological regulator of the Arf-p53 pathway in vivo. 52 Consistently, Kobayashi and Srour reported that Dmp1 regulates hematopoietic stem cell function under both steady-state and stress conditions through the regulation of Arf and p21Cip1. 54
hDMP1 is a tumor suppressor in humans as well. Loss of heterozygosity (LOH) of the hDMP1 locus was found in 42% of human breast carcinomas, while that of INK4a/ARF and p53 was found in 20% and 34%, respectively.42,53 Hdm2 amplification was found in 13% of the same sample, independent of LOH for hDMP1. LOH of hDMP1 was found in mutually exclusive fashion with that of INK4a/ARF and p53 and was associated with low Ki67 index and diploid karyotype, and longer relapse-free survival (RFS), while LOH of p53 was associated with shorter survival. 42 Recent studies suggest the critical roles of oncogenic splice variants from human genomic loci in carcinogenesis.43,55–58 We found overexpression of the splice variant DMP1β in human BC primary samples and conducted clinicopathological and transgenic mouse studies focusing on DMP1β. 59
We have been working on the signaling pathways that connect oncogenic activation and tumor suppressor genes in the p53 and RB pathways in the last decade. In this review, we have chosen four molecules, cyclin E, B-Myb, Twist, and DMP1β, which are involved in mammalian cell cycle progression by regulating the RB and p53 pathways (Fig. 1), and summarized the key findings from recent publications. MMTV-driven transgenic models have been created for cyclin E and DMP1β, while the oncogenic potentials for B-Myb and Twist have been shown by laboratory studies with siRNA. All of these molecules have negative impacts on the survival of BC patients and thus can be novel biomarkers and therapeutic targets.
Cyclin E
The Cyclin E (CCNE1) gene has been mapped to the human chromosome 19q12-q13. 60 This gene encodes a variety of polypeptides with molecular weights ranging from 39 to 52 kDa. Porter and Keyomarsi 61 reported that although Cyclin E1 is subject to extensive alternative splicing, these Cyclin E mRNA variants did not account for the low molecular weight (LMW) forms of the protein observed in tumor cells. 62 They showed that the generation of these tumor-specific LMW forms of cyclin E is predominantly derived from proteolytic processing of the full-length cyclin E1. 62 Mass spectrometric analysis revealed that the full-length cyclin E, which is a 50-kDa protein found in both normal and tumor cells, is actually the elongated (EL) form (ie, the 15-amino-acid [aa] elongated variant of cyclin E; 63 Fig. 2A). The full-length EL cyclin E consists of 411 amino acids containing nuclear localization signal (NLS) at aa 24–29 and two cyclin boxes at aa 141–238 and aa 245–342. The RB-binding motif VxCxE is at aa 289–293, in the middle of the second cyclin box (Fig. 2A; the aa numbers are those in the EL form for human cyclin E1).

The structure for human cyclin E1 and Twist1 proteins. (A) The structure of the EL form of human cyclin E160,196: It consists of 411 amino acids with two central cyclin boxes responsible for RB interaction. The NLS is at aa 24–29 (RSRKRK), 197 and the VxCxE motif 198 essential for RB binding has been mapped at aa 274–278 of the molecule. Elastase cleaves FL cyclin E at aa 40 and 69. 199 (B) The structure of human Twist1: The bHLH structure essential for DNA binding is at the center of the molecule (aa 109–163). The two NLSs are located at the N-terminal domain (aa 37–40 and 73–77). 200 The Tryptophan Arginine (WR) domain essential for interaction with core EMT factors 201 has been mapped to the C-terminus.
The second mammalian E cyclin, cyclin E2, was identified in 1998 64 and is largely considered as being functionally redundant with cyclin E1 (Figs. 1, 2A, and 4).21,65 The Cyclin E2 gene (CCNE2) has been mapped to the human chromosome at 8q22.1 and thus is different from the locus of CCNE1. The cyclin E1 and E2 proteins display high sequence similarity (69.3% in Homo sapiens). They have been considered to be functionally redundant because cyclin E1-/- and E2-/- double-knockout mice are embryonic lethal, while cyclin E1-/- and E2-/- single-knockout mice have primarily normal phenotypes. 65 However, cyclins E1 and E2 are regulated by distinct TFs and miRNAs. Moreover, the expression patterns of cyclins E1 and E2 are not always linked with cancer, and this discordance indicates that there are underlying functional differences between the two proteins. 65 We will call cyclin E1 as “cyclin E” hereafter in this review as most of the research on cyclin E has focused on cyclin E1 than cyclin E2.
Cell cycle deregulation of cyclin E expression is common in tumor cells, leading to constitutive cyclin E expression and cyclin E/Cdk2 activity throughout the cell cycle. Indeed, aberrant overexpression and activity of cyclin E have been reported with a variety of human cancers (eg, carcinomas of the breast, ovary, lung, stomach, and uterus),28,66–69 which contribute to the oncogenic process. The Cyclin E locus is amplified by an eightfold increase, but the transcript is overexpressed by a 64-fold increase in a subset of BC cell lines (MDA-MB-157). 70 Cyclin E gene amplification was observed in as many as 15 different tumor types, ie, carcinomas of the breast, bladder, ovary, uterine cervix, endometrium, gastrointestinal tract, gall bladder, and sarcomas. 71 Whole-genome characterization of chemoresistant ovarian cancer (OC) revealed that CCNE1 amplification was common in the primary disease. 69 Decreased degradation via the ubiquitin-proteasome pathway is another mechanism leading to cyclin E overexpression. Consistently, genes for the F-box proteins that target cyclin E for polyubiquitination/degradation have been reported to be mutated in human cancers. 72
Cyclin E not only plays critical roles in G1–S progression by binding and stimulating Cdk2 but also accelerates tumor development by increasing genomic instability 73 and influencing EMT through Slug, 74 a transcriptional repressor known to control EMT and promote cancer invasion/metastasis. At G1/S transition, cyclin E/Cdk2 mediates phosphorylation of Slug at Ser 54/104, which results in its ubiquitination and proteasomal degradation. As a consequence, nonphosphorylatable Slug is stabilized at G1/S transition, leading to downregulation of DNA synthesis and checkpoint-related proteins, such as TOP1, DNA Ligase IV, and Rad17. This, in turn, reduces cell proliferation and contributes to genomic instability. Thus, the cyclin E/Cdk2-Slug pathway has multiple roles in cancer progression by controlling G1-S progression, EMT, and genomic stability.
One of the most important mechanisms for the deregulation of cyclin E activity is the generation of LMW isoforms following cleavage of full-length cyclin E, 62 as described earlier (Fig. 2A). In BCs, cyclin E is cleaved to LMW fragments of 33-45 kDa by elastase and calpain 2 (Fig. 2A).62,75 Of note, the LMW cyclin E binds more tightly to CDK2 than the wild type, which leads to increased CDK2 activity and decreased sensitivity to inhibition by p21CIP1 or p27KIP1.76,77 Cells with LMW cyclin E expression have genomic instability due to premature activation of CDC25C and shortening of the length of M phase of the cell cycle, 78 which is related to the resistance to tamoxifen. 79 Clinically, the expression of LMW forms of cyclin E strongly correlates with decreased survival in patients with BC 80 and thus are desirable molecular targets for cancer therapy.
Creation of transgenic/knock-in/knockout mouse models has become essential to evaluate the biological activities for oncogene/tumor suppressor gene targets.81,82 Akli et al studied the oncogenic potential of LMW cyclin E by creating MMTV-driven transgenic mice and showed that those overexpressing LMW proteins had increased incidence of mammary tumors and distant metastasis as compared with mice expressing full-length cyclin E. 83 To test the requirement for Cdk2 in LMW cyclin E-mediated mammary tumorigenesis, Doung et al 80 created transgenic mice that expressed LMW cyclin E in a Cdk2-deficient background. They found that mammary gland development proceeded relatively normally in mice lacking Cdk2, indicating that the kinase activity was largely dispensable for this process. Interestingly, Cdk2 depletion induced cell death in LMW cyclin E overexpressing human BC cell lines, indicating that Cdk2 is required in LMW cyclin E-mediated mammary tumorigenesis. Therefore, human BCs overexpressing LMW cyclin E are primary candidates for anti-CDK2 therapy. 80
Cyclin E as a molecular marker for BC
Intact cyclin E protein has been detected by immunohistochemistry (IHC) in formalin-fixed, paraffin-embedded (FFPE) tissue, 84 and LMW forms of cyclin E have been detected by Western blotting of freshly frozen BC specimen. 85 Elevated levels of the cyclin E protein have been fairly consistently associated with a poor prognosis in BC. The levels of total and LMW cyclin E in tumor tissue, as measured by Western blotting, strongly correlated with shorter survival in BC patients. 85 Wang and Shao 86 conducted a meta-analysis of 12 published studies with 2,534 BC patients. The combined hazard ratios (HRs) for RFS were 2.32 and 1.72 in univariate and multivariate analyses, respectively. Gao et al 87 also conducted a meta-analysis of 7,759 BC patients from 23 studies and evaluated the correlation between cyclin E overexpression and survival in BC. Combined HRs suggested that cyclin E overexpression had an unfavorable impact on overall survival (OS; HR = 1.30) and BC-specific survival (BCSS; HR = 1.48), but not on disease-free survival (DFS; HR = 1.11), in patients with BC. Significantly, risks were found among stage I–II BC patients (HR = 1.75). In conclusion, high level of cyclin E is an independent prognostic factor to OS/BCSS of BC patients. 87
Substantially higher poor prognostic value has been reported for cyclin E when both the full-length and LMW cyclin E are considered together by Western blotting. 85 However, there are two issues to be resolved to apply their results to large prospective studies that should be conducted in multiple institutions because (1) Western blotting is relatively time-consuming to conduct in a pathology laboratory as a laboratory test for an individual and (2) the antibody used in the study by Keyomarsi et al 85 cannot be applied to immunohistochemical studies with FFPE samples. Monoclonal antibodies that work with cyclin E IHC are needed for repeating prognostic studies in FFPE archived tissue and finally to make their use in routine clinical practice possible.
Cyclin E as a therapeutic target for BC
Doung et al 80 studied the signaling pathways deregulated by LMW cyclin E in BC patients to identify pharmaceutical agents to effectively target this pathway. Ectopic LMW cyclin E expression in nontumorigenic human mammary epithelial cells (HMECs) was sufficient to generate xenografts with greater tumorigenic potential than full-length cyclin E. However, cyclin E mutants unable to interact with CDK2 protected HMECs from tumor development. When HMECs were cultured in Matrigel, LMW cyclin E mediated aberrant acinar morphogenesis, including enlargement of acinar structures and formation of multiacinar complexes. Of note, the B-Raf-ERK1/2-mTOR pathway was activated in LMW cyclin E-expressing patient samples and activation of this pathway was associated with poor disease-specific survival. 80 Combination treatment using roscovitine (CDK2/5/7 inhibitor) plus either rapamycin (mTOR inhibitor) or sorafenib (a pan-kinase inhibitor targeting B-Raf) effectively prevented aberrant acinar formation in LMW cyclin E-expressing cells by inducing G1/S cell cycle arrest. 80 Akli et al reported that LMW cyclin E requires CDK2 activity to induce mammary tumor formation by disrupting acinar development. 88 In conclusion, the B-Raf-ERK1/2-mTOR signaling pathway is aberrantly activated in BC with LMW cyclin E, which can be suppressed by combination treatment with roscovitine plus either rapamycin or sorafenib.
Mittendorf et al studied the interaction between HER2/neu and cyclin E in BC. 89 Decreased HER2-mediated signaling resulted in decreased expression of cyclin E, particularly the LMW isoforms, which resulted in decreased cyclin E-associated kinase activity and cell proliferation. The monoclonal antibody to HER2 trastuzumab (Herceptin®) reduced cyclin E expression in BC cells in vivo. They also found synergistic effects between trastuzumab and the Cdk2 inhibitor roscovitine in HER2(+) BC. Together, HER2-mediated signaling increases LMW cyclin E expression, which, in turn, deregulates G1-S progression of the cell cycle. 89 As LMW cyclin E expression is associated with aggressive BC, it is a desirable target for molecular therapy.84,89,90
Cyclin E overexpression and resistance to Cdk4/6 inhibitors
Human cancers that lack p16 INK4a or overexpressing cyclin D1 (eg, due to CCND1 gene amplification, promoter activation, or decreased degradation of the protein) 11 have been expected to have increased sensitivity to CDK4/6 inhibitors,31,91 while normal cells are relatively resistant assuming that most cancer cells have become addicted to the functional loss of RB. 92 However, amplifications of CCND1 or CDKN2a loss did not predict the response to CDK4/6 inhibitor therapy, as reported by clinical trials with BC patients,91,93 indicating that genetic alterations that inactivate the RB pathway may not serve as a biomarker in selecting patients who should receive CDK4/6 inhibition therapy. Although RB deficiency will cause striking resistance to CDK4/6 inhibitors, mutations affecting other cell cycle regulators, such as amplification of CCNE1, loss of p21 CIP1 /p27 KIP1 , or activation of CDK2 through different mechanisms, would bypass the requirement of tumor cells for the CDK4/6 activity that will result in the accumulation of hypophosphorylated RB.14,94–98
In OC, Konecny et al 99 showed that cell lines with higher expression of RB were associated with lower IC50 values and thus are more sensitive to the CDK4/6 inhibitor PD0332991 (P = 0.007), whereas cell lines with high expression of p16INK4a or cyclin E1 were associated with higher IC50 values and thus are more resistant to the chemical (both P < 0.001). 99 Then, a cluster diagram of the 40 OC cell lines was developed using cell cycle markers, such as RB, CDKN2a, and CCNE1. When cell lines were ordered from low IC50 values to high IC50 values, it becomes clear that sensitive OC cell lines showed high RB expression but lower p16INK4a or cyclin E1 expression; the levels of other cell cycle regulators were not strongly correlated with in vitro sensitivity to PD0332991. 99 Using the OVCAR3 cell line (RB loss and CCNE1 amplification), Taylor-Harding et al depleted endogenous cyclin E1 by short-hairpin RNA (shRNA) and showed that downregulation of cyclin E1 increased PD0332991 sensitivity and led to a near complete loss of anchorage-independent growth in a different OC cell line HEY. 100 These studies indicate that the cyclin E1 overexpression is associated with PD0332991 resistance in OC cells. Although the results of comparable studies have not been reported in BC with PD0332991 or other CDK4/6 inhibitors, it is highly possible that cyclin E1 overexpression is as important as RB deficiency as a mechanism of resistance to these novel chemotherapeutic agents. Further research is required to establish the role of cyclin E in the treatment of BC with CDK4/6 inhibitors as they have been recently approved for BC therapy.101–103
B-Myb
The c-Myb proto-oncogene was first identified as the mammalian homolog of v-Myb, which is the oncogene for the avian myeloblastosis and E26 retroviruses causing acute leukemia in birds.104–107 In humans, the Myb family comprises A-Myb, B-Myb, and c-Myb; the former two genes had been isolated during screening of human cDNA libraries at low stringency. 108 Each family member recognizes and binds to the same DNA consensus sequence [PyAAC(G/T)G] to transactivate gene expression; however, tissue-specific expression and protein–protein interactions with unique cofactors suggest that distinct biological roles exist for each Myb family protein.109,110 Although Dmp1 has Myb-like repeats, it does not bind to consensus sequences of Myb proteins, but binds to some Ets consensus with GGAT core, and thus is different. 119 Myb proteins are encoded in the genomes of both plants and animals and control a variety of processes from flavonoid production to cellular proliferation.110,111 In contrast to vertebrates, invertebrates contain only one Myb protein. The Drosophila Myb is phylogenetically and functionally equivalent to vertebrate B-Myb, suggesting that B-Myb is the most ancient family member. 112
B-Myb is homologous to c-Myb in the DNA-binding domain, and the transcripts have been detected in the majority of cell lines and tissues (Figs. 3A and 4). Like c-Myb, B-Myb acts as a TF.106,107,110 The B-Myb gene is highly expressed in embryonic stem (ES) cells developing mammalian tissues and adult hematopoietic precursors, suggesting that its expression is linked to cell proliferation. The expression is barely detectable in G0 and is induced at the G1/S transition of the cell cycle. It is phosphorylated by cyclin A/Cdk2 during S phase, which activates the protein (Fig. 4).113,114 It is considered that B-Myb phosphorylation interferes with corepressor binding and enhances B-Myb transcriptional activity. It should be noted that while cyclin/Cdk2-directed phosphorylation activates B-Myb, it also causes accelerated protein turnover. Being a classical E2F target (Fig. 1), 13 B-Myb has been shown to promote S phase entry, DNA replication, and transcriptional activation of genes, such as cyclin B1, Plk1, and Bub1, which are essential for G2/M phase progression and mitosis (Fig. 4). 115 B-Myb also co-targets genes regulated by Oct4, Sox2, and Nanog that are significantly associated with stem cell differentiation, embryonic development, and epigenetic control. 115 Cyclin D1 interacts with the B-Myb transcriptional domain, quenching B-Myb transactivation by interfering with CBP/p300 in a Cdk-independent fashion.11,116–118 The mechanism is very close to the inhibition of another Myb-like protein Dmp1 activity in a Cdk-independent fashion.119,120 When cells exit quiescence in response to growth factors, they generate a burst of cyclin D1, which is required for further progression in the cell cycle. It has been proposed that cyclin D1 maintains B-Myb in a repressed state until cyclin D1 is degraded in late G1 where phosphorylation of B-Myb by mitotic cyclin A/Cdk2 switches on transcription of B-Myb-target genes in S to G2/M phases of the cell cycle (Fig. 4).113,114,116–118
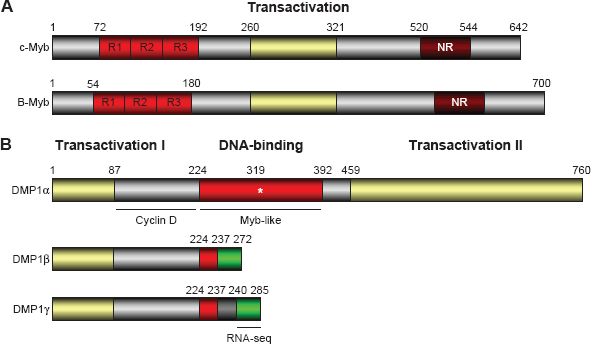
The structure for human B-MYB and DMP1β. (A) The structures of chicken c-Myb and human B-MYB: Myb family proteins (A-Myb, B-Myb, and c-Myb) have three tandem Myb-like repeats for DNA binding at the N-terminus, the central transactivation domain, and the C-terminal regulatory domain. The C-terminal domain has a negative-regulatory domain. The three tandem Myb-like repeats are also found in DMP1.119,120 Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2; 202 however, the role of the Myb family proteins in telomere maintenance has not been studied. (B) The structure of the human DMP1α and DMP1β/γ splice variants: The DMP1α has a central DNA-binding domain with three tandem Myb-like repeats with flanking transactivation domains. 119 It is an authentic TF with tumor suppressive activity, which is regulated by D-type cyclins in Cdk-independent fashion. Conversely, the DMP1β and γ splice variants lack most of the DNA-binding domain and the C-terminal transactivation domain. 172 The green box shows a unique amino acid sequence found in DMP1 β and γ. The DMP1 isoform-specific antibody (RAB) was developed using an epitope sequence (NH2-LWTPKKGHTFKLWLSKYC-COOH). 59 The green area specific to DMP1 p/y has been used in the RNA-seq analysis of the public database GSE58135.
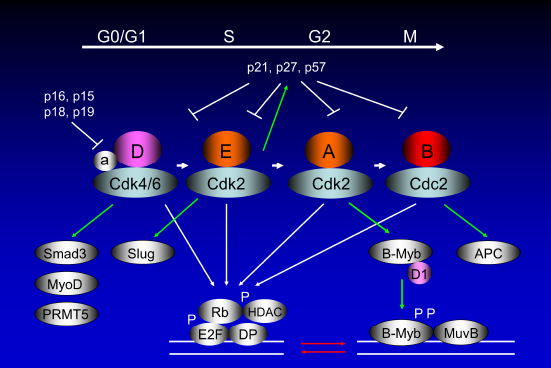
Mammalian cell cycle progression involving cyclin E and B-Myb. Mammalian cell cycle consists of alternating DNA synthetic (S) and mitotic (M) phases, separated by two gap phases (G1 and G2).13,16 The cells respond to extracellular mitogens and antiproliferative cytokines from the time they exit mitosis until they reach the restriction point, after which they can complete the cell division cycle in the absence of extracellular growth factors. Cyclin D-dependent kinases accumulate in response to mitogenic signals and initiate phosphorylation of Rb, a process that is completed by cyclin E-Cdk2. Once cells enter the S phase, cyclin E is degraded and cyclin A enters into complexes with Cdk2. Ink4 proteins oppose the activities of the Cdk4/6, whereas Cip/Kip proteins inhibit all of the enzymatic activities of cyclin D/Cdk4/6, cyclin E/Cdk2, cyclin A/Cdk2 (or Cdc2), and cyclin B/Cdc2. 14 The B-Myb expression is barely detectable in G0 but is induced at the G1/S transition of the cell cycle. It is phosphorylated by cyclin A/Cdk2 during the S phase, which activates the protein. B-Myb phosphorylation interferes with corepressor binding and thus enhances its transcriptional activity.113,114 It should be noted that while cyclin A/Cdk2-directed phosphorylation activates B-Myb, it also causes accelerated protein turnover. Being a classical E2F target (Fig. 1), B-Myb has been shown to promote S phase entry, DNA replication, and transcriptional activation of genes, such as cyclin B1, Plk1, and Bub1, which are essential for G2/M phase progression and mitosis. Cyclin D1 interacts with the B-Myb transcriptional domain, quenching B-Myb transactivation by interfering with CBP/p300 in a Cdk-independent fashion.11,116–118 B-Myb collaborates with MuvB and FOXM1 for transcription at G2-M phase of the cell cycle. 107 It should be noted that numerous genes have both E2F- and B-Myb-binding sites in their promoters, where these two proteins synergize as TFs (red arrows).107,115,203 Cyclin D/Cdk binding is different from other cyclin/Cdk binding as assembly factors (a) 204 are required for their specific binding. The cyclin D/Cdk complex has other targets than Rb, namely, Smad3, MyoD, and PRMT5.205–207 Cyclin E/Cdk2 phosphorylates p27Kip1 for proteasomal degradation, which is essential for the cells to enter the S phase of the cell cycle. 15 The major target for phosphorylation by cyclin B/Cdc2 is anaphase-promoting complex (APC).
The physiological functions of B-Myb have been studied through a number of gene knockout or depletion experiments. Although the heterozygous mutant mice were healthy, the homozygous mutants died at an early stage of development, around E4.5–E6.5. 121 In vitro culture of blastocyst indicated that B-Myb is required for inner cell mass formation. 121 Consistent with the important role of B-Myb in early embryonic development, only B-Myb among Myb family members was expressed in embryonic stem cells. These results indicate that each of the three members of the Myb gene family plays a distinct role during development. 121 García et al later created conditional knockout mice for B-Myb and demonstrated that in B-Myb-/-, mouse embryonic fibroblasts (MEFs) showed decreased growth that correlated with aberrant nuclear DNA replication. 122
Depletion of B-Myb resulted in delayed transit through G2/M, severe mitotic spindle, and centrosome defects and in polyploidy. 123 Moreover, many euploid ES cells that were transiently deficient in B-Myb become aneuploid and thus were no longer viable. 123 Downregulation of B-Myb in ES cells decreased Oct4 RNA and protein, while its overexpression modestly upregulated Pou5f1 gene expression. 123 The coordinated changes in B-Myb and Oct4 expression were attributed to the ability of B-Myb to modulate Pou5f1 gene promoter activity. Ultimately, the loss of B-Myb and associated loss of Oct4 led to an increase in early markers of differentiation prior to the activation of caspase-mediated programed cell death. Thus, appropriate B-Myb expression is critical for the maintenance of chromosomal stability and pluripotency of ES cells for differentiation. 123 Lorvellec et al later showed that B-Myb ablation led to stalling of replication forks and superactivation of replication factories that resulted in disorganization of the replication program and an increase in double-strand breaks. 124 These effects were partly due to aberrant transcriptional regulation of cell cycle proliferation factors, namely, c-Myc and FoxM1, which dictate normal S phase progression. In conclusion, during the S phase, B-Myb plays a critical role in facilitating the accurate progression of replication, thereby protecting the cells from genomic damage in ES cells. 124
B-Myb and BC
The human B-MYB chromosomal locus, 20q13, is amplified and/or overexpressed in a variety of cancers, including breast, prostate, liver, and ovarian carcinomas associated with poor prognosis. 110 B-Myb is an important marker of poor outcome in embryonal tumors of the central nervous system (medulloblastomas, neuroblastoma, ependymoblastoma, etc.). 125 Two nonsynonymous B-Myb germline variants (rs2070235 and rs11556379) causing a serine-to-glycine or isoleucine-to-methionine amino acid change (S427G and I624M) were linked to a decrease in overall cancer risk for neuroblastomas, chronic myelogenous leukemia, and colon cancers in a combined data set of cases and controls. 126 Of note, these polymorphisms are commonly found in 10–50% of human beings. 126 Surprisingly, the former polymorphism (S427G) was linked to the increased risk of basal-like BC, 127 although the mechanism remained unknown. To study the role of B-Myb in BC, they analyzed the expression of B-Myb in different BC subtypes and found an obvious association between the B-Myb expression and BC subtypes: it was highest in basal-like BC, followed by HER2+/ER– and luminal B, and lowest in normal-like and luminal A, 127 indicating that B-Myb expression was a sign of aggressive BC. Importantly, BCs overexpressing B-Myb was associated with significantly shortened OS in locally treated luminal A, luminal B, and HER2+/ER– BCs; BC survival was also shortened in RFS. 127 Cells ectopically expressing wild-type B-Myb (or the S427G variant) showed increased sensitivity to DNA topoisomerase IIα inhibitors (doxorubicin and etoposide) in human breast epithelial cell lines. In addition, microarray analyses identified many G2/M genes as being induced in B-Myb overexpressing cells. These results indicate that B-Myb is involved in cell cycle control and that its dysregulation contributes to an increased sensitivity to a specific class of chemotherapeutic agents. Hence, it is important to examine the B-Myb gene in BC for the determination of disease risk and treatment guidance. 127
Dedić Plavetić et al 128 analyzed five proliferation markers: Ki67, aurora-A kinase, survivin, B-Myb, and cyclin B1 by IHC in BC. Tissue microarrays were also conducted in 215 BC tumor samples. Statistically significant prognostic influence of aurora-A kinase, survivin, and B-Myb expression levels on shortened OS and DFS was found, and the influence of cyclin B1 expression level on DFS was also found. A multivariate analysis showed that survivin and B-Myb expression were independent prognostic factors for OS and DFS in BC patients. 128
Tao et al 129 showed that increased B-Myb expression was associated with BC progression and the protein levels were significantly elevated in BC metastases. High B-Myb levels also predicted shorter OS of BC patients, consistent with the preceding study. 128 B-Myb stimulated transcription of target genes that promoted entry into the S and M phases of the cell cycle, cell proliferation, migration, and invasion in BC. In summary, B-Myb has a critical role in both cell cycle progression and tumorigenesis and might serve as a novel molecular target in the diagnosis and/or treatment of human BC. The same group also reported the role of B-Myb in EMT and cancer metastasis. 130 They found that siRNA-mediated depletion of B-Myb expression restored E-cadherin expression and cell-cell junction formation in BC cells, suppressing cell invasion, anchorage-independent growth, and tumor formation. Conversely forced B-Myb expression decreased the expression of the E-cadherin but instead increased the mesenchymal markers for BC. They also showed that B-Myb upregulated the expression of the key EMT regulator Snail, which, in turn, mediated EMT activation and cell invasion by B-Myb. 130 In summary, B-Myb is a critical regulator for cell cycle progression and accelerates BC metastasis by upregulating Snail expression.
Twist
The TWIST1 gene located on human chromosome 7q21.2 encodes a basic helix-loop-helix (bHLH) TF that plays essential roles in organogenesis.131–135 The name “Twist” was based on the observations that in the absence of the Twist1 gene, Drosophila embryos failed to form the ventral furrow at gastrulation, lacked mesoderm and all internal organs, and eventually died at the end of embryogenesis with a twisted phenotype.131,132,136 Twist1 shows its developmental function by controlling cell migration and tissue reorganization and is a master regulator for blastula gastrulation, mesoderm differentiation, somatic muscle patterning, and specification during early embryogenesis. 137 Twist1 is evolutionarily conserved from invertebrates to humans, 135 and mutations in the Twist1 gene have been known to cause Saethre-Chotzen syndrome (SCS) in humans.138–140 SCS is a rare autosomal-dominant hereditary disorder showing craniosynostosis, facial asymmetry, ptosis of eyelids, widely spaced eyes, and broad nasal bridge, proving its essential role in normal human development. The closely related Twist2 gene (Dermo1) has been mapped onto human chromosome 2q37.3 and has been considered to inhibit osteoblast maturation and maintain cells in a preosteoblast phenotype; 141 however, not much is known about its role in carcinogenesis.
An early study indicated the oncogenic role of Twist in carcinogenesis by suppressing ARF expression (Fig. 1). 142 A functional screen for cDNAs that could counteract with the proapoptotic effects of the Myc oncogene identified two related bHLH family members, Twist (Twist1) and Dermo1 (Twist2). Both of these proteins inhibited oncogene- and p53-dependent cell death. Twist expression bypassed p53-induced cell cycle arrest, which correlated with an ability of Twist to interfere with activation of p53 target genes in response to DNA damage. Importantly, Twist affected p53 indirectly through inactivation of the ARF-MDM2-p53 self-autonomous tumor suppressor pathway. 142 Twist is overexpressed in a variety of cancers, including breast, lung, prostate, and gastric carcinomas (Fig. 5). 143 Consistent with the role as a potential oncoprotein, Twist expression promoted colony formation of adenovirus E1A/Ras-transformed MEFs in soft agar. Furthermore, Twist was highly expressed in half of human rhabdomyosarcomas. Thus, Twist may play multiple roles in the development of rhabdomyosarcomas, inhibiting terminal differentiation by interfering with the ARF-p53 tumor suppressor pathway (Fig. 5). 142
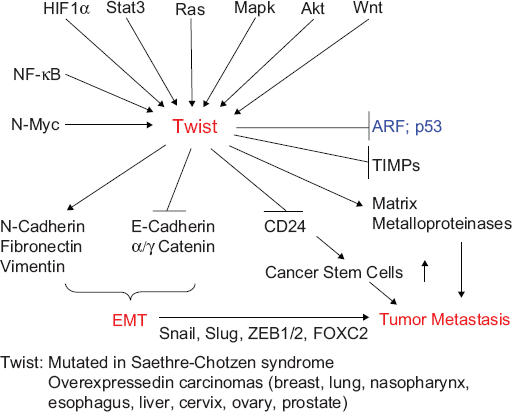
Signaling cascades through Twist and their roles in carcinogenesis. The bHLH TF Twist1 induces EMT and escape to oncogenic hyperproliferation-induced fail-safe program, facilitating the intravasation of cancer cells in the systemic circulation and their dissemination to distant organs. 208 It is overexpressed in a variety of cancers, including breast, lung, prostate, and gastric carcinomas. 143 The nuclear protein Twist is activated by a variety of signal transduction pathways, including N-Myc, NF-κB, HIF1α, 209 signal transducer and activator of transcription 3 (STAT3), Ras, mitogen-activated protein kinase, Akt,143,156,158 and Wnt signaling. Activated Twist, in turn, upregulates N-cadherin, fibronectin, and vimentin and downregulates E-cadherin and α/γ catenin, which are the hallmarks of EMT. EMT programs are orchestrated by a set of pleiotropically acting TFs–-including Twist, Slug, Snail, ZEB1, ZEB2, FOXC2, and others–-which organize entrance into a mesenchymal state by suppressing the expression of epithelial markers and inducing expression of other markers associated with the mesenchymal state. 210 Twist plays an important role in some other physiological processes involved in metastasis, such as angiogenesis, invadopodia, extravasation, and chromosomal instability by activating matrix metalloproteinases, and inhibits tissue inhibitor of metalloproteinases. Twist also protects cancer cells from apoptotic cell death through inactivation of the ARF-p53 pathway. In addition, Twist increases BC stem cells by transcriptional repression of CD24 to increase CD44-high and CD24-low cell population 150 and confers chemotherapy resistance. 158
The N-Myc oncogene (on chromosome 2p24.3 in humans) amplification is a frequent event in neuroblastoma and is strongly correlated with advanced disease stage and treatment failure. N-Myc overexpression promotes both cell proliferation and p53-dependent apoptosis by sensitizing cells to a variety of insults. By performing a pan-genomic cDNA microarray analysis, Valsesia-Wittmann et al 144 demonstrated that human Twist was constantly overexpressed in N-Myc-amplified neuroblastomas. N-Myc induces cell proliferation, whereas Twist inhibits the apoptotic response through the Arf-p53 pathway that is triggered by c-Myc overexpression in normal cells. This cooperation allows incipient cancer cells to override the intracellular fail-safe mechanisms, permitting tumor initiation and progression. Their findings provide a mechanistic explanation for the rarity of p53 mutations in neuroblastomas, highlighting the oncogenic cooperation of two crucial regulators of embryogenesis (ie, N-Myc and Twist), supporting the hypothesis that neuroblastoma originates from a developmental defect (Fig. 5).
Twist in cancer progression: EMT, invasion, metastasis, and stem cell-ness
Later studies demonstrated the role of Twist in cancer progression and metastasis (Fig. 5). The Twist family bHLH proteins are involved in EMT, 145 a process in which epithelial cells lose their cell polarity and cell-cell adhesion, and obtain migratory and invasive properties of mesenchymal cells.146,147 They function by activating several target genes that promote cellular dedifferentiation and mobility. Cancer metastasis consists of several steps: EMT, intravascular invasion, transportation via circulation, extravasation, proliferation at the secondary site, and formation of metastatic lesions. 148 Twist enhances the ability of cells within a primary tumor to undergo EMT, allowing tumor cells to migrate away from the primary tumor, enter the lymphatic system and/or blood stream, and settle into secondary tumor sites. 149
Consistent with the role of Twist in EMT in cancer, Twist is thought to promote the cancer stem cell phenotype 150 (Fig. 5) and contribute to hormone therapy resistance.151,152 Mani et al showed that the induction of EMT by Twist or Snail in mammary epithelial cells increases stem cell-rich population with high CD44 and low CD24 expressions, while isolated mammary epithelial stem-like cells express EMT-inducing factors, including Twist, Snail, Slug, ZEB1, ZEB2, and FOXC2, and other EMT marker genes (Fig. 5). 153 Another study showed that Twist increases BC stem cells by transcriptional repression of CD24 to increase CD44-high and CD24-low cell population. 150 Furthermore, expression of Twist or Snail in HER2-transformed mammary epithelial cells also facilitates EMT and generates cancer stem cells that efficiently form mammospheres, soft agar colonies, and tumors. 153 Moreover, Battula et al 154 further demonstrated that Twist-, Snail-, TGFβ-induced EMT could convert HMECs to mesenchymal stem-like cells (MSCs) with the capacity to differentiate into multiple cell types, including osteoblasts, adipocytes, and chondrocytes. They also demonstrated that these EMT-derived cells have the ability to migrate toward tumor cells and wound sites as mesenchymal stem cells do. 154 Together, the EMT-derived cells are similar to MSCs in gene expression, multilineage differentiation, and the ability to migrate toward tumor cells and wound sites.
Consistent with these reports, overexpression of Twist is common in metastatic carcinomas,134,155,156 including aggressive and metastatic forms of BCs. 157 In a search for key regulators of metastasis in a murine model, Yang et al 149 found that the Twist plays an essential role in metastasis. Suppression of Twist expression in highly metastatic 4T1 mammary carcinoma cells specifically inhibited their ability to metastasize from the mammary gland to the lung. 149 Ectopic expression of Twist resulted in (1) loss of epithelial markers, such as E-cadherin, α/γ-catenins, and cell-to-cell adhesion; (2) activation of mesenchymal markers, such as N-cadherin, fibronectin, and vimentin; and (3) induction of cell motility, suggesting that Twist contributes to metastasis by promoting EMT. In human BCs, high level of Twist expression correlated with invasive lobular carcinoma, a highly infiltrating BC associated with loss of E-cadherin expression. These results established a mechanistic link between Twist, EMT, and tumor metastasis. 149
To understand the molecular basis for metastasis accelerated by Twist, Cheng et al 158 systematically selected for highly invasive cells from BC cell lines MCF7 and MDA-MB-453. Interestingly, Twist and AKT2 were found to be elevated in the invasive BC cells compared with the parental controls. Ectopic expression and downregulation of Twist resulted in significant increase and reduction, respectively, in AKT2 expression. Silencing AKT2 decreased Twist-driven migration, invasion, and paclitaxel resistance, suggesting that AKT2 is a critical downstream target for Twist. Finally, they observed a correlation of elevated Twist and AKT2 expression in the late-stage BCs as opposed to 13% in the early stage. In summary, Twist is a positive transcriptional regulator of AKT2 expression and Twist-AKT2 signaling is involved in promoting invasion and shortened survival of BC.
Twist, miRNAs, and tumor invasion/metastasis
The miRNAs are short noncoding RNAs of 20–25 nucleotides that regulate gene expression posttranscriptionally, mainly by binding to a specific sequence of the 3′ untranslated region (UTR) of target genes.8,159 Since the first report on the clinical relevance of miRNAs in cancer, many miRNAs have been demonstrated to act as oncogenes, while others are tumor suppressors. 160 Ma et al showed that microRNA-10b (miR-10b) is highly expressed in metastatic BC cells and positively regulates cell migration and invasion, using a combination of mouse and human cells. 160 Overexpression of miR-10b in nonmetastatic BC initiated robust invasion and metastasis. Expression of miR-10b is induced by Twist that bound directly to the putative promoter, which, in turn, inhibited the translation of the mRNA encoding homeobox D10, resulting in an increased expression of a prometastatic gene RhoC to increase cell invasion and metastasis of BC. 160
The mammalian Twist1 3′UTRs are highly conserved and contain a number of potential regulatory elements, including miRNA-binding sites. Nairismägi et al 161 analyzed the translational regulation of Twist using luciferase reporter assays in a variety of cell lines and found that miR-145a-5p, miR-151–5p, and a combination of these were capable of repressing Twist translation, dependent on the presence of the predicted target sites in the 3′UTR. Furthermore, the repression was sensitive to locked-nucleic acid-modified miRNA antagonists, resulting in decreased migratory potential of MEFs. Understanding the in vivo mechanisms for Twist regulation might open up a possibility for therapeutic interference by gene-specific therapies.
Twist as a molecular marker for BC
The prognostic value of Twist has been studied by tissue microarray or IHC, alone or in combination with other EMT markers, such as Snail, Slug, Zeb1, 162 or with other molecular markers (SRC-1 and gelatinase), for BC.163,164 Twist expression in BC was associated with large tumor size, Ki67, HER2, and VEGF expression; negativity of ER/PR and E-cadherin; and thus poor prognosis. 143 Expression of Twist led to dramatic changes in cellular morphology, with increased proliferation, migration/invasion, and expression of EMT-related biomarkers. 143 Consistent with these findings, Twist-expressing BC was associated with lymph node metastasis and thus had negative impacts on OS, or DFS, of BC.162–165
Twist-directed cancer therapy
In addition to the role of Twist in ARF regulation, EMT, and cancer stem cell-ness, Twist expression is associated with multidrug resistance, such as taxol and vincristine, two microtubule-targeting anticancer drugs in nasopharyngeal, bladder, ovarian, and prostate cancers.133,166,167 Although Twist is highly expressed in the mesoderm-derived embryonic mesenchyme, it is primarily expressed in relatively quiescent adult stem cells located in mesoderm-derived mesenchymal tissues. 133 Thus, Twist is an attractive therapeutic target for metastatic BC. Depletion of TWIST by siRNA caused upregulation of E-cadherin,168,169 suppressed EMT, tumor invasion, and metastasis,148,170 as predicted by mouse studies. 149 Therefore, systemic administration of a Twist inhibitor could have a significant impact on Twist-overexpressing cancer cells with minimal side effects. 170 Interestingly, Twist is one of the major transcriptional targets for NF-κB responsible for antagonizing chemotherapy-induced apoptosis, suggesting an important role in NF-κB-mediated cell survival and chemoresistance. 171 Together, Twist is a promising target for molecular therapy for cancer.
DMP1β
The hDMP1 locus on chromosome 7q21 consists of 18 exons that encode three different splicing variants (α, β, and γ) with different biological activities.43,59,172,173 This locus is quite unique in that it produces both tumor suppressive and oncogenic gene products from one locus through alternative splicing. The former transcript was designated as DMP1α, while the other transcript was named as DMP1β (Fig. 3B). The biology of the third transcript, DMP1γ, is currently unknown. The DMP1β/γ proteins lack most of the Myb-like repeats responsible for DNA-binding and the entire C-terminal transactivation domains found in DMP1α and thus do not act as a TF. 172 DMP1β blocks differentiation for CD13 expression and stimulates proliferation during PMA-induced differentiation U937 cells to macrophages, while DMP1y had little effect.172,174 DMP1β and γ did not activate the ARF promoter, whereas only the former resulted in a dose-dependent inhibition of DMP1α-induced transactivation of the ARF promoter. 173 Ectopic expression of DMP1β reduced endogenous ARF mRNA levels in human fibroblasts. Mechanistically, they showed that DMP1β might interact with DMP1α to antagonize its function in vitro through DNA-binding assays and in cells by the close proximity of DMP1α/β in the nucleus. 173 The DMP1 mRNA levels were reduced in acute myeloid leukemic samples as compared to normal granulocytes. Treatment of acute promyelocytic leukemic patient samples with all-trans retinoic acid promoted differentiation to granulocytes and restored DMP1 transcripts to normal granulocyte levels. Thus, the DMP1β/α ratios were tightly regulated in hematopoietic cells; DMP1β antagonizes DMP1α's transcriptional activation of the ARF promoter, resulting in cellular proliferation. 173
As DMP1α is a critical mediator of BC (mammary tumor) suppression in humans and mice,42,45,175 185 we studied the role of the DMP1β/γ splice variants in mammary oncogenesis. Total RNAs were isolated from both tumors and adjacent neighbor tissue of 46 BC patients, and quantitative Reverse Transcriptase–-Polymerase Chain Reaction (qRT-PCR) was conducted for DMP1. 59 The DMP1β/α isoform mRNA ratio was higher in 14 of 46 BC samples (~30%) than their neighbor pathologically normal tissues, while DMP1γ/α isoform ratio was higher in only 3 of 20 tumors examined (15%). 59
We then analyzed the publicly available database at GSE58135 with the RNA sequencing (RNA-seq) technique to study different BC specimens that consist of 84 primary BCs and 51 adjacent uninvolved breast tissues (Fig. 6A and 6B). We analyzed DMP1β+γ signals as a whole as all the DMP1γ readouts are included in DMP1β (Fig. 6A and 6B; the results for DMP1β only study is shown in Ref. 59). We found that the average DMP1β+γ mRNA levels were higher in both ER+HER2– and triple-negative breast cancer (TNBC) than those in uninvolved tissues, as shown by the Student's t analyses (P = 0.0029 and 0.0052, respectively; Fig. 6C). The modes for DMP1β+γ signals were higher in BC samples than those of uninvolved neighbor tissues (95 vs. 50 in ER+/HER2– BC; 105 vs. 60 in TNBC; Fig. 6A and 6B). Overall, significantly increased expression of human DMP1β+γ mRNA (ie, 90 hits or higher) was observed in 22/42 (52.4%) of ER+/HER2– BC and 31/42 (73.8%) of TNBC patients (Fig. 6A and 6B), which is consistent with the percentage of high DMP1β protein expression in IHC stated later.
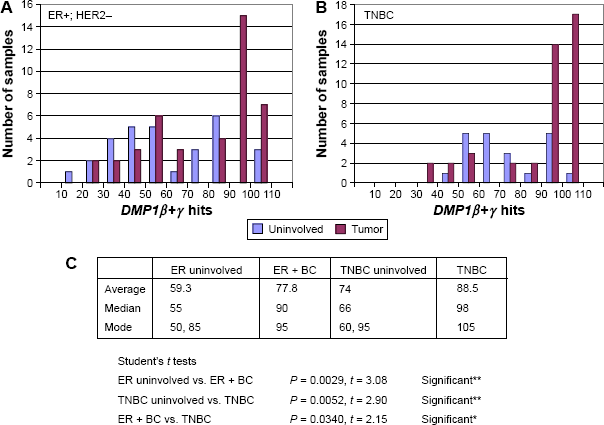
Analyses of the public database GSE58135 for BC by RNA-seq. (A) and (B) The data were obtained from the analyses of RNA-seq data of 42 estrogen receptor-positive (ER+) and HER2-negative (ER+/HER2–) BC primary tumors, 30 uninvolved breast tissue samples that were adjacent to ER+/HER2– primary tumors, 42 different TNBC primary tumors, and 21 uninvolved breast tissue samples that were adjacent to TNBC primary tumors (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58135). The x-axis shows the number of hits for DMP1β+γ and the y-axis shows the number of patients. The data were normalized by the length and abundance of the transcripts.59,211 (C) Statistical analyses for the number of DMP1β+γ readings (ie, blast alignments and hits). The DMP1β+γ readings are higher in BC than in uninvolved tissues, as shown by the Student's t analyses (P < 0.0001 in all BC; P = 0.0029 in ER+/HER2- BC; and P = 0.0052 in TNBC). The DMP1β+γ readings are higher in TNBC than ER+/HER2- BC (P = 0.034), which is consistent with the association of DMP1β with poor prognosis of BC. The median hit is higher in BC than uninvolved tissue (90 vs. 55 in ER+; 98 vs. 66 in TN BC). There are two modes (peaks) in uninvolved tissue (50 and 85 in ER+/HER2– uninvolved; 60 and 95 in TN BC uninvolved), but there is only one in BC (95 in ER+/HER2–; 105 in TNBC). Significantly increased expression of human DMP1β+γ mRNA (ie, 90 hits or higher) was found in 20/42 (47.6%) of ER+/HER2– BC and 31/42 (73.8%) of TNBC. The second peaks in uninvolved tissue are possibly from precancerous breast tissues where tumor cells are emerging.
Next, we studied whether DMP1β protein expression was increased in BC by raising an antibody specific to DMP1β (RAB). 59 Of note, RAB detected only DMP1β, but not γ, possibly because of posttranslational modifications of the antigenic epitope in DMP1γ. 59 Using the RAB antibody, we performed IHC with paraffin-embedded tumor tissues from 63 BC patients. The data indicate that 35 of 63 (56%) breast tumors were highly stained (3+ to 2+) with the RAB antibody relatively to the surrounding breast epithelial tissues without involvement (Fig. 7A, left).
We then conducted Kaplan-Meier survival analysis based on strong vs. weak DMP1β staining intensity. The patients with high DMP1β staining in the tumors relapsed earlier than those with low DMP1β staining (P = 0.0050; x 2 = 7.8653; Fig. 7B). There was no correlation between DMP1β protein expression and LOH of the locus, suggesting that these two events occurred independent of each other. Taken together, our data indicate that the DMP1β protein is frequently overexpressed in BCs and has negative impact on patients’ survival.
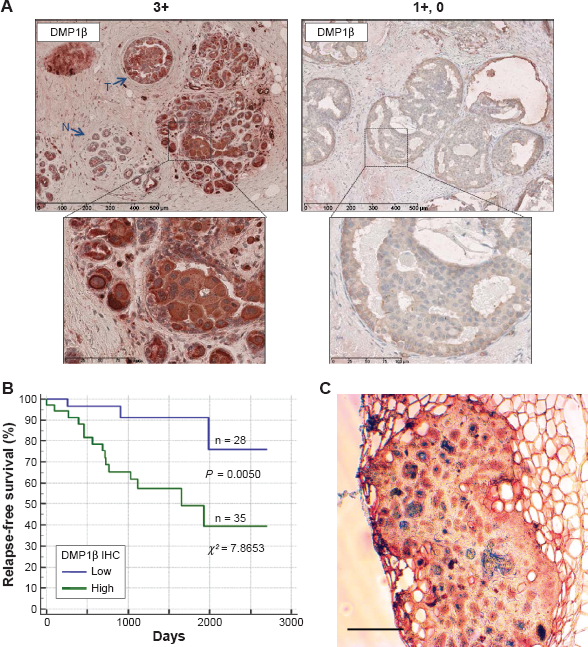
DMP1β IHC in human BC. (A) Representative images of DMP1β IHC staining from two BC patients (Patient #1: high DMP1β expression; Patient #2: low DMP1β expression). 59 A total of 63 human breast tumors were stained with DMP1β-specific antibody, RAB. DMP1β staining was significantly higher in the tumor tissues in Patient #1 compared to surrounding normal tissues. The scale bar indicates 100 μm. (B) A Kaplan–Meier RFS curve was graphed based on high versus low DMP1β protein intensity. 59 Patients with significantly higher DMP1β (high) staining in tumors compared to the surrounding normal tissue had significantly shorter relapse than the those with tumors that show undetectable DMP1β (low) in their tumor tissue (P = 0.0050; x 2 = 7.8653). (C) Mammary tumor tissue from an MMTV-DMP1β V5His female mouse doubly stained for DMP1β (peroxidase; red) and cytokeratin 14 (alkaline phosphatase; blue). The majority of tumor cells were positive for both proteins, suggesting transdifferentiation of mammary tumor cells to adenosquamous carcinoma. The scale bar indicates 100 μm.
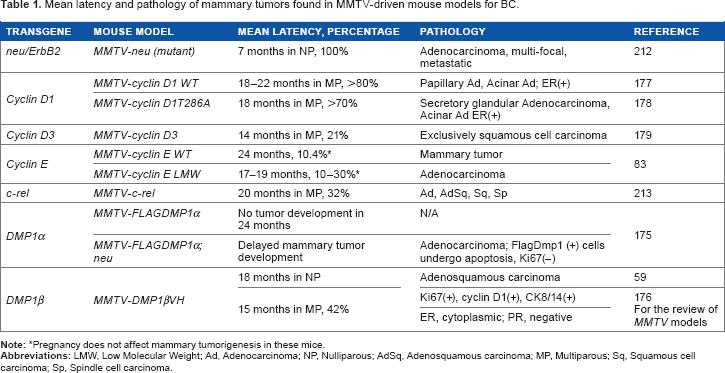
The oncogenic activity of DMP1β in mammary gland epithelial cells was demonstrated in vivo through the creation of MMTV-DMP1β V5His transgenic mouse lines. 59 The transgenic females developed mammary tumors with a mean latency of 16 months (42% of multiparous females; 18 months in nonparous and 15 months in multiparous). The onset of mammary tumor development in MMTV-DMP1β-transgenic females was earlier than those of MMTV-cyclin D1 (177, 178), D3 (179), E (83), or c-Rel (213) mice (18–24 months in parous females), but later than MMTV-neu (mutant) mice (7 months; Table 1; ref. 212). Moreover, the frequency of tumor development was 42% in the MMTV-DMP1β model, while it was only 10–30% in many other MMTV models, indicating that this is a faithful model for BC found in women older than 40 years as 12-month-old mice correspond to 40-year-old humans. IHC analyses demonstrated that the tumor cells were expressed in the proliferation markers Ki67 and cyclin D1 in DMP1β-transgenic mice. 59 The mammary tumors were also positive for cytokeratin 8/14, suggesting transdifferentiation of adenocarcinoma to adenosquamous carcinoma (Fig. 7C). 59 Although adenosquamous carcinomas are infrequent in human BC, they were found in the mammary gland of cyclin D1 and cyclin D3-transgenic mice.176–179 Together, it is possible that DMP1β induces mammary tumors through upregulation of cyclin D1 and/or D3, the mechanism that should be pursued in the future. We propose that these transgenic mice are faithful mouse models for highly proliferative BC with adenosquamous differentiation.
Mean latency and pathology of mammary tumors found in MMTV-driven mouse models for BC.
Pregnancy does not affect mammary tumorigenesis in these mice.
Conclusive Remarks
We have reviewed the publications on the four novel biomarkers for BC–-cyclin E, B-Myb, Twist, and DMP1β. All of these molecules are associated with poor clinical outcomes of BC patients, and at least the three of them (cyclin E, B-Myb, and Twist) have been shown to play critical roles in EMT and tumor metastasis. In case of cyclin E and DMP1β, transgenic expression of the protein under the control of the MMTV promoter led to mammary tumor development in vivo. Although transgenic models have not been created for B-Myb or Twist expression in mammary epithelial cells, the oncogenic role of these molecules are evident judging from their roles in cell cycle progression, the impact on the ARF-p53 pathway, EMT, and stem cell-ness of BC. Creation of transgenic mouse models, either constitutive or drug-inducible/de-inducible, will be helpful to directly demonstrate the roles of these molecules in mammary carcinogenesis and/or metastasis in vivo. The role of DMP1β in EMT/cancer metastasis should be investigated in the near future. Large multi-institutional studies with IHC with specific antibodies are needed to establish the roles of these molecular as novel biomarkers for BC. Identification of molecules that associate with these proteins and characterization of upstream/downstream signaling cascades will also be helpful to discover the drugs that inhibit the activity of these oncogenic molecules for BC therapy.
Recently, the use of CDK4/6 inhibitors has been approved in the therapy of BC.101–103 Clinical trials have shown that these inhibitors are efficacious in BC with INK4a deletion; however, those with RB deficiency or Cyclin E amplification/overexpression were resistant to CDK4/6 inhibitors in OC. 99 As it is unrealistic to express the RB gene in all tumor cells that lack RB, it will be more reasonable to downregulate the overexpressed cyclin E protein, possibly through interference of translation, acceleration of protein degradation, or gene/protein depletion by siRNA to overcome the therapy resistance. Thus, mechanistic studies on cyclin E regulation in tumor cells are very important. Over the past decade, more than 20 siRNA therapeutics have been developed for a variety of disorders, including cancer, virus infection, and genetic disorders. 180 Like other biological drugs, RNA interference-based therapeutics often require a delivery vehicle to transport them to the targeted cells. Thus, the clinical advancement of numerous siRNA drugs has relied on the development of its carriers. Advancements in bioengineering and nanotechnology for improved delivery and release of the siRNA are expected.
Accumulating data indicate the negative impact of B-Myb, Twist, and DMP1β in survival of BC. As B-Myb and DMP1β are direct E2F targets and Twist is a repressor of the INK4a/ARF locus associated with EMT, it is possible that overexpression of some of these proteins (and also other molecules that accelerate cell cycle progression–-they can be novel E2F targets) is associated with CDK4/6 inhibitor resistance in BC. Thus, it will be critical to find such molecular markers for clinical application of CDK4/6 inhibitors in BC.
Author Contributions
Both the authors contributed to the conceptual design, writing the text, creating figures, and collecting the literature publications. All authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
We thank all the members of Dr. Inoue's laboratory for sharing research findings on BC. We are grateful to Dr. Ali Mallakin who conducted the RNA-seq analysis of the BC database.