
Abstract
Pancreatic ductal adenocarcinoma (PDAC) constitutes 90% of pancreatic cancers. PDAC is a complex and devastating disease with only 1%–3% survival rate in five years after the second stage. Treatment of PDAC is complicated due to the tumor microenvironment, changing cell behaviors to the mesenchymal type, altered drug delivery, and drug resistance. Considering that pancreatic cancer shows early invasion and metastasis, critical research is needed to explore different aspects of the disease, such as elaboration of biomarkers, specific signaling pathways, and gene aberration. In this review, we highlight the biomarkers, the fundamental signaling pathways, and their importance in targeted drug delivery for pancreatic cancers.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is characterized by a series of molecular aberrations. 1 Due to the heterogeneity and the complex nature, it is hard to diagnose and treat this malignancy, which has only a 1%–3% survival rate in five years after the second stage. 2 In most cases, diagnosis occurs in the later stages, with a well-developed, dense, desmoplastic stroma and metastasis to other organs. The spreading of the disease from the pancreas to multiple distant sites renders major surgery impossible. The complexity of the disease manifests as different, patient-specific, aberrant biochemical pathways and makes the treatment challenging. The dense extracellular matrix (desmoplasia) in PDAC leads to early development of hypoxia, expression of inflammatory cytokines and other extracellular components, and epithelial-to-mesenchymal transition (EMT). All of these incriminating factors make drug delivery complicated, resulting in drug resistance and disease relapse. 3
Altered gene expression patterns and mutations are frequently observed in PDAC. 1 Gene expression microarray analysis has identified the following three main subtypes of PDAC: classical, quasimesenchymal, and exocrine like. The classical PDAC cells (BxPC3 and CaPan-2) have the characteristic epithelial-like genes, while the quasimesenchymal cells (Panc-1 and MiaPaCa-2) express mesenchymal features. Exocrine-like primary tumor cells overexpress digestive enzymes. 4 For example, tissue microarray analysis detected the expressions of ABCC3 and TLR2 in AsPC-1, CaPan-1, HPAFII, PSN-1, and SU86.86 pancreatic cancer cell lines. ABCC3 is an ATP-binding cassette mostly observed in the tumor tissues of pancreatic cancer and may be used for cell surface-targeted imaging and delivery of therapeutics. 5 Discovery of other biomarkers and aberrant biochemical pathways (contributing to tumorigenicity) has made tremendous progress in recent years. Despite the considerable research and clinical studies, PDAC is still a lethal disease. In this review article, we summarize the biomarkers of PDAC and recent developments of targeting several pathways for treating the disease.
Epidermal Growth Factor Receptor
Epidermal growth factor receptor (EGFR), a transmembrane glycoprotein of the EGFR family, is overexpressed in 40%–70% of patient samples with pancreatic cancer.1,6 The ErbB, also known as the human EGFR-1 (HER-1), belongs to the EGFR family. The glycoprotein EGFR has an intracellular tyrosine kinase domain, a transmembrane domain, and an extracellular domain for ligand binding. Interactions of the tumor growth factor-α and EGF with the extracellular domain lead to dimerization and autophosphorylation of EFGR protein, producing downstream signal transduction. Activation of the EGFR kinase stimulates the following two signaling pathways: RAS-RAF-mitogen-activated ERK-activating kinase (MEK)-mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)-PTEN-Akt-mTOR-GSK3 (Fig. 1).7,8
Signaling pathways stimulated by the activation of EGFR kinase.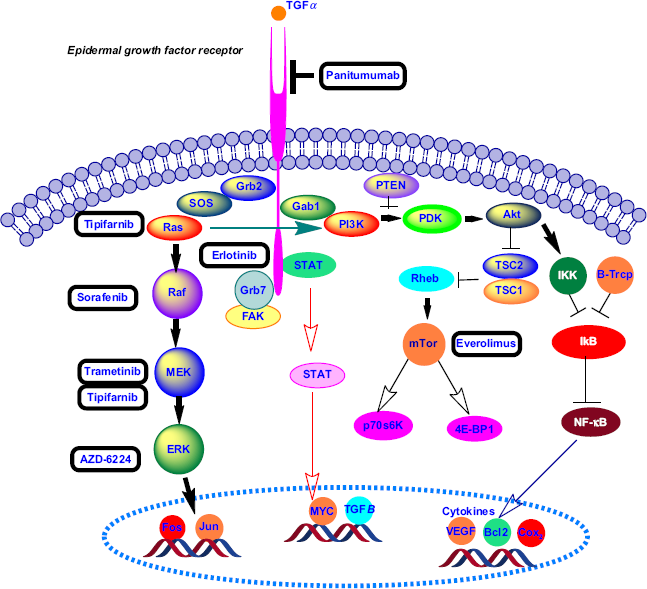
The anticancer drugs erlotinib and gefitinib inhibit the autophosphorylation of EFGR tyrosine kinase by competing with adenosine triphosphate in the intracellular domain. 7 The US Food and Drug Administration (FDA) has approved erlotinib as a combination therapy (with gemcitabine) for PDAC. Boeck et al 9 evaluated the overexpression of EGFR in tumor tissues treated with erlotinib from 181 phase III randomized patients by immunohistochemistry (49% showed EGFR overexpression). Cardnell et al reported that EMT leads to resistance to EGFR inhibitors and metastatic progression of PDAC. 10 Recently, researchers have discovered the role of Na+/H+ exchanger protein NHE1 in promoting EGFR signaling pathway and pancreatic cancer metastasis. The coadministration of cariporide (an NEH1 inhibitor) with erlotinib results in a decreased three-dimensional colony growth and invasion for both classical (BxPC3 and CaPan-2) and quasimesenchymal (Panc-1 and MiaPaCa-2) pancreatic cancer cell lines. 11 Anti-EGFR monoclonal antibodies (eg, cetuximab and panitumumab) inhibit receptor dimerization at the extracellular domain. In a recent phase II clinical study, radiotherapy along with cetuximab increased radiosensitivity in locally advanced pancreatic cancer. 12
Kirsten Rat Sarcoma Viral Oncogene
Kirsten rat sarcoma viral (KRAS) oncogene is a GTPase protein belonging to the RAS gene family. 13 In 1982, the mutated human RAS gene was found to be activated in cancer. 14 The KRAS proto-oncogene point mutation occurs in 75%–95% of PDAC. 1 The most common mutation is the replacement of glycine with aspartate at position 12 (KrasG12D). KRAS in pancreatic cancer is characterized by the mutation type, allelic ratio, and tumor subtype.15,16 Tumors with high dependency on KRAS might have poor prognosis. 4
KRAS oncogene mutation activates the P21 RAS protein and a series of signaling pathways.
17
The RAS protein is located on the inner surface of the cell membrane and binds to guanosine triphosphate (GTP)/guanosine diphosphate (GDP). In the presence of RAS mutation, GTPase cannot undergo transition from the GTP (active) form to GDP (inactive) form, and RAS remains in a permanently active state, resulting in a cascade of downstream activation.
13
Figure 2 depicts the RAS protein regulation GTP/GDP cycle. Prenylation of the RAS protein increases its capability to interact with cell membrane and endoplasmic reticulum (ER) compartments via the hydrophobic C terminus.
18
Farnesyltransferase and geranylgeranyltransferase I, respectively, attach the farnesyl (15 carbon) and geranylgeranyl (20 carbon) isoprenoid lipids to the cysteine residue of RAS protein with the C terminus of CAAX (C: cysteine, A: aliphatic amino acids, and X: usually serine or methionine).14,18 To inhibit the RAS protein, a farnesyltransferase inhibitor for posttranslational prenylation Tipifarnib (R115777) was investigated in conjunction with gemcitabine in a double-blinded phase III clinical study on advanced pancreatic cancer. However, the results did not show any statistically significant clinical benefit over gemcitabine and placebo.
19
The lack of increased efficacy may be due to the presence of other RAS isoforms (such as non-farnesylated RAS) or the RAS-independent activity of tipifarnib.17,19
RAS protein regulation through the GTP/GDP cycle.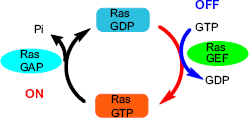
Pao et al 20 investigated KRAS mutation and the development of drug resistance to monotherapy by EGFR inhibitors (erlotinib and gefitinib) in non-small lung carcinoma. Cotreatment of locally advanced pancreatic cancer with erlotinib and gemcitabine did not significantly increase survival, even though 60% of the patients harbored EGFR expression. 21 Moreover, in a phase II clinical study, treatment with gemcitabine along with cetuximab (an anti-EGFR monoclonal antibody) was not more effective than gemcitabine alone. 22 Lee et al 23 suggested an alternate mechanism for EGFR signaling in KRAS-mutated pancreatic cancer cells that does not follow the canonical MAPK pathway. Moreover, inactivation of Akt might happen as a result of treatment with an EGFR inhibitor, such as erlotinib. 24 Three major signaling pathways, PI3K-3-phosphoinositide-dependent protein kinase-1-Akt, Raf-Mek-Erk, and Ral-GEFs, are affected by the KRAS oncogene in PDAC.4,25,26 The PI3K-3-phosphoinositide-dependent protein kinase-1 downstream pathway is mostly dominant in Kras-driven PDAC.26,27 Inhibition of tumor growth was demonstrated by blocking and deletion of Pdk-1 in the PI3K pathway in a Kras-engineered mouse model. 26 Collins et al 28 reported a mouse model of on and off Kras oncogene that developed metastatic PDAC. Inhibition of the MEK-ERK pathway using AZD-6224 in combination with glycosphingolipid synthesis inhibitor 1-Phenyl-2-decanoyl-amino-3-morpholino-1-propanol (PDMP) induced apoptosis in human pancreatic cancer. 29 Recently, Lindberg et al ruled out the effect of EGFR and HER-2 signaling pathways on the growth of patient-derived PDAC xenograft (PDX) using mice bearing wild-type and mutant Kras alleles. Coadmin-istration of panitumumab (anti-EGFR antibody) and trastu-zumab (anti-HRE2 antibody) synergistically enhanced the anticancer effect of trametinib (an MEK inhibitor) in PDX mouse models. 1 Khvalevsky et al developed the biodegradable polymer matrix Local Drug EluteR (LODER) to encapsulate Kras G12D siRNA. LODER drug eluter inhibited tumor growth by decreasing the Kras expression in an orthotopic mouse model of PDAC. 30
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases of the metzincin superfamily and degrade extracellular matrix. Therefore, they have a significant role in tissue remodeling and tumor progression in pancreatic cancer. Overexpression of MMP-1 (collagenase), MMP-2 (gelatinases-A), MMP-7 (matrilysin), MMP-9 (gelatinase-B), MMP-10, MMP-11 (stromelysin), and MMP-13 (collagenase) is observed in pancreatic cancer.31–33
Since MMPs play a key role in altering cell behavior, inhibition of MMPs is an attractive approach for anticancer therapy. Small synthetic metalloproteinase inhibitors showed promising results in preclinical studies but failed in phase III clinical trials due to lack of specificity. 34 We recently reported MMP-9-triggered release of the anticancer drug gemcitabine from liposomes. The liposomes presented a layer of polyethylene glycol on the surface for long circulation and accumulation in the tumor by the enhanced permeation and retention effect. At the tumor site, the enhanced concentration of the reducing agent glutathione reductively removed the polyethylene glycol layer and exposed the substrate peptides toward MMP-9-mediated hydrolysis. The loss of liposomal structural integrity led to the rapid release of encapsulated gemcitabine and reduction in xenograft pancreatic tumor volume in mice. 35 Munshi et al 36 reported that increased collagen leads to the overexpression of MMP-14 (MT1-MMP) in the desmoplastic regions of pancreatic cancer, causing tumor progression and gemcitabine resistance. Srivastava et al 37 reported the inhibition of MMP-2, −7, −9, and −12 by epigallocatechin-3-gallate (extracted from green tea) in vitro and xenograft mouse model of pancreatic cancer.
Receptor for Advanced Glycation Endproducts
The membrane-associated receptor for advanced glycation end-products (RAGE) belongs to immunoglobulin-like receptor family. RAGE is present in normal cells, such as epithelial cells, neurons, smooth muscle cells, and hepatocytes. The expression is upregulated in cancers and diverse types of diseases, including diabetes, Alzheimer's, osteoarthritis, and cardiovascular. 38 Several signaling cascades (eg, PI3K-Akt, MAPK, and small GTPase) are activated upon stimulation by binding of the ligands S100P, S100A4, and S100A6 to the RAGE. Overexpression of the ligands S100P and S100A6 are reported in pancreatic cancer. In a recent study, the administration of 5-methyl cromolyn (an S100 inhibitor) resulted in the reduction of tumor growth and metastasis in an orthotopic mouse model of PDAC. 39
Nuclear Factor Kappa B
The nuclear transcription factor kappa B (NF-kB) belongs to the Rel/NF-kB protein and has significant roles in targeting genes for encoding cytokines, cell growth, cell molecule adhesion, apoptosis, and inflammatory responses. 40 Overactivation of NF-kB pathway is observed in 70% of pancreatic cancer cell lines. 41 In most cases, the noncanonical NF-kB pathway overexpression is present in PDAC. 42 Small molecule inhibitors for NF-kB have not yet progressed to the clinical trials. However, several researchers have studied the inhibitory effects of curcumin (extracted from turmeric) on the expression of NF-kB using in vitro and in vivo models of pancreatic cancer.43,44 Kurzrock et al 45 reported the inhibition of NF-kB and reduced toxicity for advanced pancreatic cancer patients treated with 8 g of oral curcumin (phase II clinical trial).
Mammalian Target of Rapamycin
Mammalian target of rapamycin (mTOR) is a serine/threonine-associated PI3K signaling pathway responsible for cell proliferation, growth, and survival. The mTOR pathway is deregulated in several cancers, including PDAC. 46 Moreover, the mTOR pathway activation has been observed in pancreatic cancer stem cells (PCSCs). 47 The FDA has approved an mTOR inhibitor (Afinitor) to treat subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) and renal cancer carcinoma. Morran et al 48 demonstrated that the FDA-approved mTOR inhibitor rapamycin along with gemcitabine decreased the tumor size and proliferation in a PTEN-deficient, mouse strain with KRasG21D mutation (KC model) of pancreatic cancer. Coadministration of rapamycin and the PI3K inhibitor LY294002 impeded the proliferation and growth of PCSCs by blocking the PI3K-mTOR signaling pathway. 49
Proto-oncogene Serine/Threonine-protein Kinase
Proto-oncogene serine/threonine-protein kinase (PIM) proteins belong to serine/threonine kinase family and are upregulated in several tumors, eg, sarcoma, hepatocellular cancer, prostate cancer, and PDAC. Specifically, PIM1 and PIM3 are overexpressed in pancreatic cancer. 50 Moreover, hypoxia and Kras oncogene regulate the PIM proteins. The PIM regulates several signaling pathways in cell cycle regulation and apoptosis. The shRNA-mediated knockdown of PIM1 in MIAPaCa-2 and Capan-1 pancreatic cancer cell lines revealed that the PIM1 protein plays a significant role in anchorage-dependent and anchorage-independent growth, invasion, and radioresistance for pancreatic cancer cells. 51
V-MYC Avian Myelocytomatosis Viral Oncogene
V-MYC avian myelocytomatosis viral oncogene (MYC) protein is a transcriptional factor and has an important role in genetic and epigenetic regulations of PDAC. 52 The overexpression of Myc was observed in 32% of primary and 29% of metastatic pancreatic tumors. 53 Myc accelerates metabolism and proliferation of tumor cells and angiogenesis. 52 Zhou et al 54 demonstrated the expression of Myc in multipotent progenitor cells during differentiation into the exocrine and endocrine cells in pancreatic organogenesis. Impeding the c-Myc in pdx1+ multipotent progenitor cells resulted in altered differentiation and reduced proliferation of exocrine and endocrine pancreatic cells in a mouse model. 55 Several signaling pathways, such as PI3K-Akt, RAS-MAPK, cyclin-dependent kinase 2, and NF-kB, have a role in posttranslational alteration of Myc. 52
Platelet-Activating Factor
Platelet-activating factor (PAF) is involved in the phospholipid-regulating MAPK signaling pathway. PAF overexpression in pancreatic cancer leads to cell proliferation and tumorigenesis. Jun et al 56 demonstrated that the PAF ectopic activation of the MAPK signaling occurred via the activation of LAM TOR3 pathway, causing neoplasia in pancreatic cancer.
Cell Surface Antigen CD109
CD109, a glycophosphatidylinositol-anchored glycoprotein, was recognized as a cell surface antigen on some normal hematopoietic and metpoietic tumor cells. 57 CD109 engages in the EGF signaling in SK-MG-1 glioblastoma cells. 58 The cell surface glycoprotein CD109 was identified in BxPC3 cells from primary pancreatic cancer. CD109 glycoprotein is expressed in the BxPC3, MIACaPa-2, and Panc-1 cell lines. Also, CD109 overexpression was observed in PDAC. The expression of CD109 was evaluated in normal pancreatic tissues and PDAC samples by cell-surface capture technique and immunohistochemistry. 59
PCSC Biomarkers
Surface markers of pancreatic cancer stem cells.
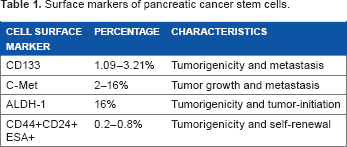
Various cellular signaling pathways, such as Notch, Wnt, and hedgehog, can facilitate the formation of stem cells in pancreatic cancer. 68 Sonic hedgehog is usually overexpressed in pancreatic tissues and PCSCs. Deregulation of sonic hedgehog pathway causes pathogenesis and desmoplasia in PDAC. 69 Cyclopamine, IPI-269609, and GDC-0449 are hedgehog inhibitors. Cyclopamine inhibits PCSCs and reduces endothelial-to-mesenchymal transition and metastasis in vitro and in vivo. 69 Moreover, expression of the cell surface biomarkers CD133 and CD44 decreases in gemcitabine-resistant cells after cyclopamine therapy. 70 Feldmann et al 71 reported that in an orthotopic xenograft model of PDAC, treatment with cyclopamine and gemcitabine decreased the expression of ALDH, resulting in reduced invasion of PDAC.
The Notch signaling pathway has important roles in cellular differentiation, apoptosis, stem cell regeneration, EMT, drug resistance, and tumorigenesis. 72 The Notch signaling pathway proteins are overexpressed in pancreatic cancer cells and PCSCs.68,73 Notch acts as the tumor suppressor in the skin and small cell carcinomas but as an oncogenic protein in pancreatic cancer. 73 Notch signaling pathway is activated through γ-secretase. The γ-secretase inhibitor MRK-003 can be used to block the Notch signaling pathway in pancreatic cancer. Coadministration of MRK-003 and gemcitabine resulted in reduced tumor size in PDAC xenograft model. 74
Another cell surface biomarker of CSCs is the tyrosine kinase C-Met. Cabozantinib, a C-Met inhibitor, impedes sphere formation and escalates apoptosis via downregulation of C-Met, CD 133, and SOX2 in PCSCs. 75 Cotreatment of XL184 (a C-Met inhibitor) with gemcitabine or XL184 alone reduced cell proliferation and growth of PCSC in NOD-SCID mice. 76 Recent research by Singh et al 77 reported that PAK4 (p-21 activated kinase 4, serine/threonine kinase family) activates the STAT3 signaling pathway, resulting in a stemness phenotype. In addition, they demonstrated that the PAK4 overexpression in PCSCs compared to the non-CSCs is associated with chemotherapy resistance and sphere formation.
Epithelial-to-Mesenchymal Transition
Through the process of EMT, epithelial cells lose their normal characteristics, such as apical-basal polarity, cell-cell tight junctions, and transition to spindle-like, motile, and invasive mesenchymal cells. 78 EMTs can be of the following three types: Type I (embryogenesis), Type II (wound healing and organ fibrosis), and Type III (cancer). 79 In addition to embryogenesis and wound healing, 80 EMT plays pivotal roles in metastasis and drug resistance in pancreatic and other cancers. 81 Due to the EMT in pancreatic cancer, epithelial cells downregulate E-cadherin and upregulate vimentin, N-cadherin, and fibronectin. 78 For pancreatic cancer patients with EMT in the primary tumor, 75% showed metastasis to the lungs and liver. 82 Tumor microenvironmental factors, such as hypoxia, inflammatory cytokines, extracellular components, and mechanical characters contribute to EMT progression. 3 The inflammatory cytokines' transforming growth factor-β (TGFβ), tumor necrosis factor-α, interleukin-1, and interleukin-6 cause progression of EMT in PDAC. 3 The TGFβ signaling pathway can act either as a tumor suppressor or as a tumor promoter, depending on the stage of PDAC. 83 The TGFβ signaling pathway leads to apoptosis in the early phases of the tumor but in later stages contributes to tumor progression and invasion via EMT. 84
The TGFβ signaling pathway upregulates TWIST1, SNAIL1, and SNAIL2 transcription factors. 3 TGFβ pathway inhibitors, such as trabedersen (AP12009) and galunisertib (LY2157299), decreased metastasis and invasion in animal model studies and clinical trials.85,86 In contrast, the TGFβ inhibitors SB431542 and galunisertib showed opposite effects when the Panc-1 cells and normal fibroblasts (VI-38) were cocultured in a three-dimensional collagen gel. The Panc-1 cells showed rapid invasion, changes in morphology, and EMT after treatment with TGFβ inhibitors. It is possible that the secreted hepatocyte growth factor from the fibroblasts and the cancer cells (in response to TGFβ inhibitors) cause invasion and cell proliferation of the Panc-1 cells into the collagen gel. 87
MicroRNAs in Pancreatic Cancer
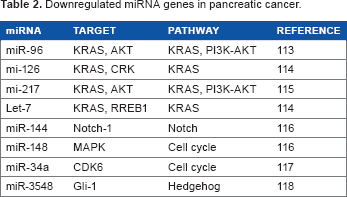
MicroRNAs (miRNAs) are small, single-stranded, noncoding, 20–25 nucleotide RNA sequences with regulatory effects on gene expressions and in several physiological and pathological processes. 88 miRNAs behave as tumor suppressors and oncogenes in pancreatic adenocarcinoma. Overexpression of the oncogene miRNAs (oncomir) increases in tumor progression, while tumor suppressors inhibit cell proliferation and induce apoptosis. 89 The miRNAs are expressed selectively in the tumor tissues 90 and inactivate the tumor suppressor genes p53, p16, and SMAD4 in pancreatic cancer. 91
The miR-21 is upregulated in pancreatic cell lines and tissue and decreases survival rate significantly. 92 The miR-21 overexpression is reported as the lesion initiator, causing tumor progression in a KRAS (G12D) mouse model. 92 The miR-155 is also upregulated in pancreatic cancer and contributes to tumor progression. Knockdown of miR-155 downregulates EGFR, KRAS, and MT1-MMP expressions, leading to inhibition of cell proliferation. 93 The upregulation of miR-221 in pancreatic cancer leads to distant metastasis and unresectable tumors. 94
Point mutation of p53 is present in 50%–70% of human pancreatic cancers. 95 The p53 facilitates transcription of a vast number of miRNAs. Stress signaling in cells induced by hypoxia and starvation upregulates p53 and activates the expressions of several genes, such as miR-107, −34a/b/c, and miR-34. The expressed miRNAs modulate apoptosis and inhibit hypoxia in PDAC.91,96 Mutation of p53 mediates transcription of miR-130b and miR-155, modifies the expressions of the corresponding target genes (ZEB1 and ZNF652), and leads to cell proliferation and invasion in several cancers.97,98 In addition, p53 mutation impairs maturation of miR-145 and miR-16-1 causing cell proliferation, invasion, and migration in PDAC. 91
Downregulated miRNA genes in pancreatic cancer.
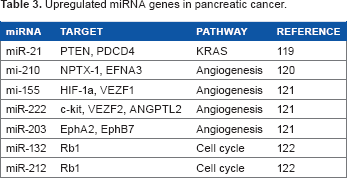
Upregulated miRNA genes in pancreatic cancer
Treatment of Pancreatic Cancer
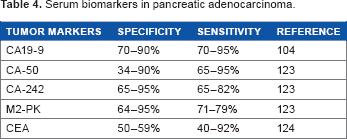
Serum biomarkers in pancreatic adenocarcinoma.
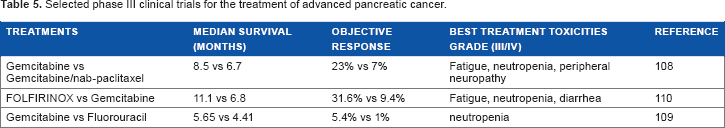
Selected phase III clinical trials for the treatment of advanced pancreatic cancer.
Conclusion
PDAC is a sequence of complex deviations at the molecular levels. 111 Cell signaling pathway alterations, pancreatic stem cells, and EMT led to resistance to conventional chemotherapy. In this review, we summarize the primary molecular changes, biomarkers, and small molecule inhibitors for blocking different pathways of PDAC. Although several inhibitors are reported for most of the molecular aberrations, extensive efforts need to be made to bring the research to the clinics. Targeted delivery reduces toxicity and enhances the efficacy of the anticancer drugs. 112 The knowledge of biomarkers and small molecule inhibitors is expected to promote further research and development of targeted therapies, alleviating the severe side effects of pancreatic cancer therapy and increasing the survival rates.
Author Contributions
Analyzed the data: FK. Wrote the first draft of the manuscript: FK. Contributed to the writing of the manuscript: SM. Agree with manuscript results and conclusions: FK and SM. Jointly developed the structure and arguments for the paper: FK and SM. Made critical revisions and approved final version: FK and SM. Both author reviewed and approved of the final manuscript.