
Abstract
Acquired resistance to targeted inhibitors remains a major, and inevitable, obstacle in the treatment of oncogene-addicted cancers. Newer-generation inhibitors may help overcome resistance mutations, and inhibitor combinations can target parallel pathways, but durable benefit to patients remains elusive in most clinical scenarios. Now, recent studies suggest a third approach may be available in some cases—exploitation of oncogene overexpression that may arise to promote resistance. Here, we discuss the importance of maintaining oncogenic signaling at “just-right” levels in cells, with too much signaling, or oncogene overdose, being potentially as detrimental as too little. This is highlighted in particular by recent studies of mutant-BRAF in melanoma and the fusion kinase nucleophosmin-anaplastic lymphoma kinase (NPM-ALK) in anaplastic large cell lymphoma. Oncogene overdose may be exploitable to prolong tumor control through intermittent dosing in some cases, and studies of acute lymphoid leukemias suggest that it may be specifically pharmacologically inducible.
Introduction
For most cancers, the hope that single-molecule targeted therapy would revolutionize treatment has been thwarted by acquired resistance. Mechanisms may be target-dependent, typically mutations or overexpression, or target-independent, such as the activation of parallel pathways. 1 Although rational strategies exist to overcome parallel signaling and resistance-conferring mutations (reviewed elsewhere2–6), the intriguing possibility of exploiting target overexpression is now also entering discussion. Here, we review the feasibility of this approach, where target upregulation can cause an overdose of oncogenic signaling that is detrimental to cancer cell survival. By exploiting the “Goldilocks principle”—the idea that even for oncogenes “just-right” levels are required—strategies such as intermittent dosing may permit prolonged tumor control in some patients.
Oncogene Addiction
Though cancer cells accumulate numerous genetic changes, deactivation of individual oncogenes often cause pronounced tumor regressions in mouse-model systems. Bernard Weinstein first used the term “oncogene addiction” in 2002 to describe these findings. 7 Strong clinical support came with the remarkable and durable efficacy of breakpoint cluster region-Ableson kinase (BCR-ABL) tyrosine kinase inhibitors (TKIs) in treating chronic myeloid leukemia (CML) without significant toxicity.8,9 Such findings revolutionized cancer drug development toward targeted therapies to exploit every tumor's perceived Achilles’ heel and ultimately driving a new paradigm of personalization of cancer care through precision medicine.6,10–12 Unfortunately, however, CML still stands more or less alone in the success of this strategy. Inhibitors targeting other driver kinases produce progression-free survival (PFS) times of typically a few months and have failed to replace frontline chemotherapy in most diseases.13–16 Even when other inhibitors have moved to the frontline, as the epidermal growth factor receptor (EGFR) inhibitor erlotinib and the anaplastic lymphoma kinase (ALK) inhibitor crizotinib have for appropriate advanced lung-cancer patients, median PFS is ~9–10 months.17,18 In TKI-treated CML by contrast, median PFS is still unknown because it has not been reached after a decade or more of follow-up.9,19–22
Newer Understandings of Oncogene Addiction
Difficulties in recapitulating the success of BCR-ABL kinase inhibitors vs. CML in other cancers have highlighted the deep gaps in our comprehension of oncogene addiction's complexities. Through recent work, however, it is now well understood that the inhibition of a single target can lead to the rewiring of signaling pathways that may rescue the activation of key downstream processes. For example, in triple-negative breast cancer, Duncan et al 23 showed how MEK/ERK inhibition promotes the degradation of MYC, leading to the expression and activation of various receptor tyrosine kinases (RTKs). These RTKs overcome inhibition of MEK2 (but not MEK1), reactivating ERK and culminating in drug resistance. Knockdown of ERK, MYC, AKT, or mTOR recapitulates this reprograming of the kinome, while preventing MYC's protea-somal degradation blocks it.23–25
Oncogenic shock
The “oncogenic shock” model provides a framework for conceptualizing such results. Briefly, while oncogenes promote proliferation and survival, they paradoxically activate signals to promote apoptosis or cell cycle arrest.26,27 This may be because of feedback repression of normal survival pathways (as above) and/or through stress imposed by increased cellular growth rates, similar to oncogene-induced senescence in nontransformed cells. 28 Either way, when an inhibitor of a driver oncogene is introduced to the system, both the growth/survival and the proapoptotic/growth-arrest signals are inhibited. In truly oncogene-addicted cells, abrupt loss of the survival signal destroys the cells before the loss of the apoptotic signal can allow them to survive and vice versa for the cells that survive.6,26,27
The oncogenic shock model, which was originally proposed nearly a decade ago, has received strong support from recent results with key clinical targeted inhibitors. In CML, the poster-child disease for oncogene addiction, for example, Asmussen et al 29 found that BCR-ABL represses normal myeloid survival pathways through feedback inhibition mediated by activated MEK, and these pathways come rushing back several hours after BCR-ABL inhibition. However, drug sensitivity is preserved because cells commit to apoptosis before rescue can occur. The story is different for BRAFV600E-driven melanoma (Fig. 1A), where inhibitors typically delay progression for only a few months. Here, the time taken for MAPK pathway rebound is a lot quicker than that for CML (2–4 hours vs. 8–24 hours).29,30 Therefore, resistance arises in mutant BRAF melanomas, as growth factor pathway restoration is established before a significant proportion of cells enter apoptosis.
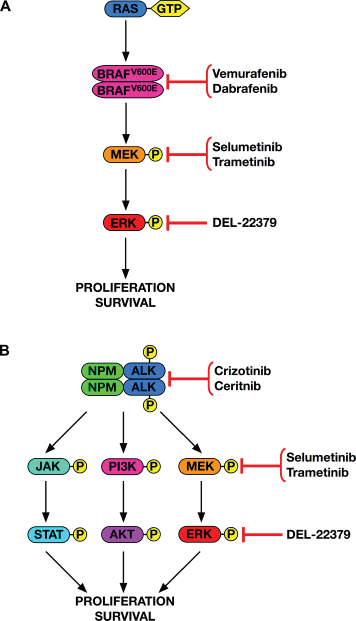
Downstream signaling and targeted pathway inhibition for mutant BRAF in melanoma (
While apoptosis precedes the restoration of oncogenic signaling through network rewiring in wild-type BCR-ABL cells, 29 complications arise in the context of resistance. For example, the BCR-ABL inhibitors imatinib, dasatinib, and nilotinib have weak off-target activity against RAF that drives RAS-dependent paradoxical BRAF and CRAF activation. 31 Because BCR-ABL is upstream of RAS activation in sensitive cells, inhibition with these agents also suppresses RAS. In drug-resistant cells with the T315I gatekeeper BCR-ABL mutation, however, RAS activity permits the drugs’ paradoxical BRAF/CRAF and downstream MEK/ERK activation (Fig. 1A). This results in dependency on this pathway, such that combining BCR-ABL inhibitors with a MEK inhibitor synergistically inhibited resistant cell growth in vitro and in vivo. 31 However, prolonged MEK inhibition can cause autocrine activation of STAT3 via fibroblast growth factor receptors and Janus kinases (JAKs), 32 supporting even further combination therapy, such as adding JAK/STAT inhibitors to the drug cocktail. Combined drug toxicities to patients, however, become a rising concern as more and more drugs are put together. Instead, what may be more practical clinically would be scheduling drug dosing intermittently, carefully pulsed to ensure target inhibition followed by withdrawal to prevent pro-survival pathway reactivation. Important work is being carried out to understand the specific timings of these antagonistic processes and their responses in different contexts based on factors like protein turnover. 6 In addition, a new approach to target the MAPK pathway—the direct inhibition of ERK with the dimerization inhibitor DEL-22379—was recently reported. 33 This drug showed ability to overcome many resistance mechanisms that thwart MEK inhibitors but has not yet been tested in BCR-ABL-driven models.
Oncogene Overdose
While extensive efforts are underway targeting signals to which cancer cells are addicted, the prospect of using these very signals to overwhelm the system is just now entering exploration. Similar to drug addicts overdosing on the very thing they require to avoid withdrawal, cancer cells may be susceptible to the induction of overdoses in oncogenic signaling. Recent studies indicate that this may be feasible in BRAFV600E-addicted melanoma, ALK-addicted T-cell lymphoma, and BCR-ABL+ B-cell acute lymphoblastic leukemia (B-ALL).
Oncogene overdose in BRAFV600E melanoma
While BRAFV600E-addicted melanomas rely on continual BRAFV600E → MEK → ERK signaling
34
(Fig. 1A), Das Thakur et al
35
found that the continual administration of vemurafenib caused resistant tumors to actually become dependent on the continued inhibition of this very pathway. Resistance arose through elevated
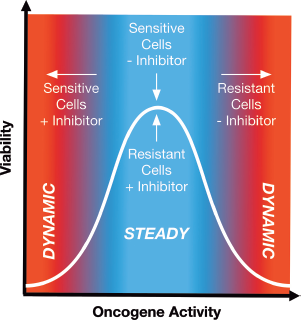
Oncogene-addicted cells can require “just the right” amount of signaling for survival. Oncogene-addicted cells constitutively express the amount of activated oncogene required for growth and proliferation. Targeted inhibition of the oncogene shuts off this signaling and shifts the cells into a dynamic state leading to death. Resistance can be achieved by increased expression and/or activation of the oncogene in question, which allows resistant cells to grow in the presence of the inhibitor originally designed to kill them. Resistance by this means is often accompanied by dependence, such that inhibitor withdrawal causes an overdose of oncogenic signaling that overwhelms the cells and also results in death.
To combat resistance to
Oncogene overdose in ALK+ ALCL
The anaplastic lymphoma kinase (ALK) is an important new therapeutic target activated through chromosomal translocations that fuse its C-terminal kinase domain to the N-terminus of various constitutively expressed proteins (Fig. 1B). Examples include t(2;5)(p23;q35) creating
We recently established, however, that overexpression of
In vivo, even stronger than the described observations in melanoma, we found that tumor engraftment of resistant cells was only seen in mice dosed with an ALK TKI (ceritinib).
56
Complete absence of engraftment in vehicle-treated mice underlines the drug dependent phenotype that these resistant cells acquired. Careful passaging of resistant cells in vitro at high confluence, however, allowed us to re-establish lines able to grow in the absence of inhibitor. ALK activity in these lines returned to baseline, and the cells were resensitized to the ALK TKIs. This is similar to observations from the above-mentioned study of melanoma, which showed resensitization through forced knockdown of
These studies of mutant BRAF melanoma and ALK+ ALCL highlight the importance of having “just the right” amount of signaling with respect to tumor survival (Fig. 2).
Goldilocks Principle
These findings fit well into the “Goldilocks principle”, the idea that certain biologic factors require precise levels to promote fitness, with either too much or too little being toxic. For example, the restoration of calcium release in heart cells, 57 the levels of oxygen administered by postcardiac arrest, 58 the amount of vitamin D in the body, 59 the redox environment of the cell with respect to oxidative stress, 60 and the levels of MeCP2 in causing Rett syndrome immune defects 61 must fall within the “Goldilocks zone.” Even a person's body mass index, which should be within the “normal” range, can be thought of in terms of the Goldilocks principle.
MAPK cascade and the Goldilocks principle
Although not usually described in these terms, RAF signaling also follows a Goldilocks paradigm, having dual roles in cell cycle progression or arrest depending on level.62,63 Studies mostly in mouse fibroblasts, for instance, showed decreases in Raf activity can promote passage through the cell cycle through activation of Cyclin D1/Cdk4 and Cyclin E-Cdk2. Too much Raf, however, can cause cell cycle arrest through the induction of the Cdk inhibitors p21CIP1 and p16INK4A.62,63 Interestingly, Raf appears to suppress its own activity to maintain levels in the Goldilocks zone. 64 In addition, further highlighting the paradoxical nature of RAS → RAF → MEK → ERK signaling, the overactivation of any step in the pathway can cause oncogene-induced senescence via replicative stress leading to the activation of p16INK4A and p19ARF tumor suppressor pathways.28,65–67 In an inducible mouse model with titratable levels of Ras, increased signaling led to senescence, while lower levels induced tumor formation. 68 Therefore, optimum tumor development also requires “just-right” levels of RAS, and indeed, this can be achieved by way of balancing autophagy.69,70 Furthermore, autophagy itself appears to follow the Goldilocks principle in tumor development. On the one hand, it allows cells to scavenge their own reserves during nutritional and ischemic stress. On the other hand, too much autophagy opposes tumor establishment and development, through nascent tumor death or the prevention of further mutations that favor progression caused by reactive oxygen species that are generated by the buildup of damaged organelles. 71
Intermittent dosing
The requirement that at least some oncogenes maintain signaling at “just-right” levels may be exploitable using intermittent dosing to delay the onset of fatal resistance (Table 1). As discussed above, pulsed TKI dosing may allow BCR-ABL and its downstream signaling cascade to be effectively turned off on-drug, without permitting the reactivation of growth factor signaling when off-drug.6,29 This can provide a window where there are only proapoptotic signals and no pro-survival signals present, enhancing cell death. Furthermore, pulsed BCR-ABL inhibition elicits the activation of the proapoptotic BIM protein with the same kinetics seen with continual exposure. 72 This approach, however, is highly specific depending on the oncogene in question. For example, while the inhibition of FLT3-ITD fusion in acute myeloid leukemia turns off oncogenic signaling, growth factor receptor signaling is restored at a faster rate upon inhibitor discontinuation, which means that there is not enough time for the induction of apoptosis before the reactivation of pro-survival signaling.6,29 It will require meticulous tweaking of dosing schedules to exploit each oncogene to bring about death before the restoration of pro-survival signaling.
Studies that favor intermittent dosing to prolong tumor control.
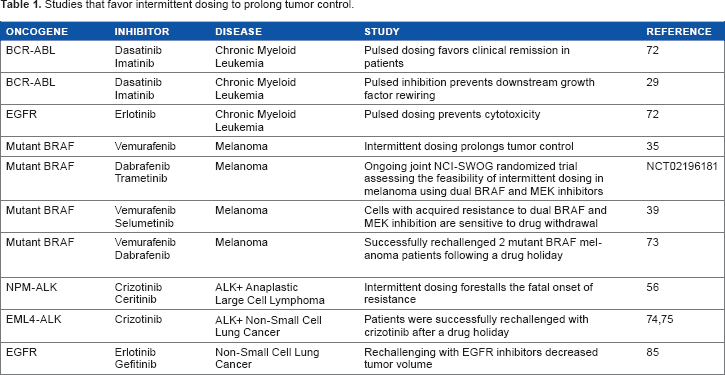
Systems in which target overexpression drives both resistance and dependence may be the most amenable to intermittent-dosing strategies. Using patient-derived BRAFV600E melanoma xenografts, Das Thakur et al
35
showed that intermittent vemurafenib dosing prolonged tumor control compared to continuous dosing. Both individualized drug interruptions based on tumor burden and up-front scheduled intermittent dosing were superior to continuous dosing in this report. Considering strategies to combat targeted-drug resistance, intermittent dosing emerges as one carrying the least amount of both expense and toxicity to patients. Therefore, the melanoma report both established the preclinical proof of principle and highlighted its potential flexibility. A small case series, meanwhile, reported successful BRAF-inhibitor rechallenge in two patients who had previously developed resistance and were then off-drug for several months, but the cause of resistance was not investigated.
73
A joint NCI-SWOG randomized trial specifically comparing intermittent vs. continuous dosing of dabrafenib and trametinib for treating
Although we found that ALK+ ALCL lines selected for TKI resistance reliably developed resistance-dependence, we have only begun exploring intermittent dosing as a therapeutic strategy. The improvement of ALK+ ALCL mouse models using genome editing with CRISPR/Cas9 should allow this possibility to be more rigorously investigated. However, two case reports have shown that patients may respond positively to rechallenge with crizotinib, lending support to the clinical investigation of discontinuous dosing with ALK TKIs.74,75
Pharmacologically Inducing Oncogene Overdose
Another potential way to exploit therapeutically oncogene overdose may be through its forced pharmacological induction. In order to investigate this possibility, the mechanism(s) underlying oncogene overdose must be well understood. In ALK+ ALCL, our knowledge is incomplete, and, as mentioned, the inhibition of downstream targets of ALK failed to rescue cell viability (unpublished observations). We are undertaking several unbiased approaches to elucidate the specific mechanisms underlying overdose in the hope that forced induction may be translatable into a therapeutic strategy.
In another lymphocyte-derived malignancy, however, both the basis for and means to induce an overdose of oncogenic signaling were recently established. Briefly, the Goldilocks principle can be thought of as applying to the positive and negative selection of developing lymphocytes. The positive selection of immature B and T cells requires their newly created B-cell receptor (BCR) or T-cell receptor (TCR) to bind test antigens with a minimum affinity to weed out weak clones not likely to be useful in immune defense. However, negative selection eliminates clones with too strong of an affinity to self-antigens, eliminating clones likely to precipitate autoimmune phenomena. In both the cases, the degree of affinity is translated into the strength of downstream signaling, with too little or too much both being potentially fatal. Therefore, lymphocytes carry a propensity in their life history to die due to the overactivation of their core survival pathways.76–79
Approximately 25% of B-ALL cases contain the chromosomal fusion BCR-ABL discussed above, which in this context mimics the constitutive activation of pre-BCR signaling.80,81 A comprehensive study by Chen et al 82 showed that BCR-ABL ALL cells express increased amounts of several immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing proteins, such as PECAM1, CD300A, and LAIR1. These ITIMs recruit several phosphatases, such as PTPN6 (also known as SHP1) and INPP5D, which down-regulate the activation of SYK, an early step in BCR signaling whose activation is normally markedly reduced in BCR-ABL ALL cells. Therefore, the authors used a small molecule inhibitor that targets the inhibitory phosphatase INPP5D to induce SYK activation. This led to the selective death of BCR-ABL+ B-ALL cells, both in vitro and in vivo. Furthermore, this was achieved regardless of the mutational status of BCR-ABL, as SYK induction also caused BCR-ABLT315I ALL cell death. This study highlights the potential of pharmacologically inducing overdose as a form of therapy.
This paradigm may also be applicable to T-ALL. A study of
Conclusions
More work is warranted to assess the impact of oncogene overdose as a general mechanism leading to cell death in the context of acquired resistance, and intermittent dosing to exploit overdose may be fruitful in prolonging tumor control in selected patients. The in vitro and in vivo work discussed above in mutant BRAF melanoma and ALK+ ALCL shows great promise for potentially implementing this treatment paradigm in a patient setting, where intermittent dosing forestalled the onset of resistance.35,56 The findings of these studies are further corroborated by clinical cases where discontinuous dosing has been highly efficacious (discussed above and summarized in Table 1). However, care must be taken when administering this regimen, as drug withdrawal prior to the onset of resistance, as well as the dose of drug initially administered, could accelerate acquired resistance.6,84 Therefore, up-front intermittent schedules must be carefully studied and established. Otherwise, individualized approaches may be better, particularly for the short term. Precise characterization of the mechanisms of oncogene overdose will shed clearer light on both the therapeutic potential and appropriate strategies of intermittent dosing and is required before any efforts to pharmacologically induce overdose can be undertaken. It is also important to note that while intermittent dosing is less toxic and expensive than most other treatment strategies, it is not a cure. The in vivo experiments in mutant BRAF melanoma show that although the forestalling of resistance is achieved via intermittent dosing, eventually the cell finds a way and resistance refractory to intermittent dosing sets in. 35 When feasible, regular tumor screening and genotyping to ascertain the mechanisms at play when tumor burden increases at each drug cycle will help clinicians stay one step ahead of tumor evolution and prevent the fatal onset of resistance.
Author Contributions
Wrote the first draft of the manuscript: ADA and JHS. Contributed to the writing of the manuscript: ADA, SSR, MJG, PP and JHS. Jointly developed the structure and arguments for the paper: ADA and JHS. Made critical revisions and approved final version: ADA, SSR, MJG, PP and JHS. All authors reviewed and approved of the final manuscript.