
Abstract
A synthetic gene encoding bovine terminal deoxynucleotidyl transferase (TdT) was generated, cloned into an expression vector and expressed in E.coli. The effects of altering culture and induction conditions on the nature of recombinant protein production were investigated. This led to the expression of active recombinant bovine TdT in E.coli. After purification and characterisation, the activity of the enzyme was assessed in a biological assay for apoptosis. The process described in this report enables the economical production of TdT for high throughput applications.
Introduction
Terminal deoxynucleotidyl transferase (TdT) is a DNA polymerase that belongs to family X of the DNA polymerase super-family. 1 The enzyme is expressed in immature pre-B, pre-T lymphoid and acute lymphoblastic leukemia cells and it is used as a marker to diagnose certain hematological malignancies.2,3 It adds nontemplated nucleotides to the variable, diversity and joining exon junctions of antibody genes during antibody gene recombination playing a vital role in the generation of antibody diversity. 4 The enzyme functions by catalyzing the addition of nucleotides to the 3’ terminus of its substrate DNA molecule and is unusual among the DNA polymerases as it does not require a template. It shows substrate preference toward a 3’ overhang, but can also add nucleotides to blunt or recessed 3’ ends and cobalt is a necessary cofactor for enzymatic activity. 5 TdT is used in many molecular biology techniques including RACE (rapid amplification of cDNA ends) to add nucleotides to the ends of cDNA molecules which can then be used as templates for PCR. 6 It is also used to add nucleotides labeled with radioactive isotopes.
TdT is the enzyme used in the TUNEL (
TdT from calf thymus gland was first purified over thirty years ago as a proteolysed form containing two peptides. Although calf thymus gland is an abundant source for TdT, the proteolysis that occurs in this tissue makes purification of single chain TdT problematic. Purification of enzyme from TdT positive cultured cell lines in the quantities required would be impractical in cost and effort. For this reason many attempts have been made to generate TdT through recombinant DNA technology. Most recombinant TdT is generated using a baculovirus based system. 8 Attempts to express TdT in a cheaper and higher yielding bacterial system have been generally unsuccessful. Less active forms of TdT, for example avian have been successfully expressed in E.coli 9 Also human TdT has been expressed in the same bacteria but at very low yields and poor purity. 10 To the authors’ knowledge there are no reports of bovine TdT being generated in E.coli. The main factors contributing to these disappointing outcomes are the intrinsic insolubility of eukaryotic proteins in prokaryotic hosts 11 and the incompatibility of codon usage in eukaryotic gene sequences to the abundance of tRNA species in bacteria. 12 This report describes the successful manipulation of these factors affecting protein expression to generate active bovine TdT in E.coli.
Materials and Methods
The nucleotide sequence for bovine TdT (NP_803461) was processed using the web-based DNA codon optimisation algorithm Upgene 13 and the codon usage of the sequence was optimised for expression in bacteria. Restriction sites were added to the 5’ (Nde I) and 3’ (BamH I) ends of the gene to facilitate the downstream cloning into an expression vector and the resulting gene sequence was synthesized by GenScript Corporation, Piscataway, New Jersey. The synthetic gene was sub-cloned from its holding vector (pUC57) into the expression vector pET19b (Novagen, La Jolla, California). This expression vector encodes a poly-histidine tag which is expressed on the N terminus of the recombinant protein to allow affinity chromatography. The insert in the recombinant plasmid pET19b_bovTdT was fully sequenced to confirm its identity and sufficient quantities of DNA were generated to transform the following E.coli host strains: BL21(DE3), BL21(DE3)pLysS, BL21(DE3) Star, Rosetta-gami2(DE3), Rosetta-gami2(DE3)pLysS, Rosetta-gamiB(DE3) and Rosetta-gamiB(DE3)pLysS. Small cultures (20 ml) of each of the host strains harbouring the recombinant plasmid pET19b_bovTdT were grown to an OD600 of 0.5 at 37 °C. Expression was induced by adding IPTG to a final concentration of 1 mM. 1 ml aliquots were removed at 0, 30, 60, 90, 120, 150 and 180 minute time points and analysed by SDS PAGE. At the end of the induction period, 1 ml of culture from each induction was processed to produce soluble and insoluble fractions which were also analysed by SDS PAGE. The whole induction process was repeated at 30 °C and at 22 °C. The yield of recombinant protein generated was assessed as was the proportion present in the soluble and insoluble fractions.
A larger induction (500 ml) was performed using the Rosetta-gamiB(DE3)pLysS strain harboring the recombinant plasmid. The cells were grown at 30 °C to an OD600 of 0.5 and induced with 1 mM IPTG for 180 minutes. Aliquots of 1 ml were taken at the 0 and 180 minute time points, soluble and insoluble extracts were generated and the samples were analysed by SDS PAGE. After confirmation of a successful induction, the entire 500 ml induced culture was processed to generate a soluble and an insoluble fraction. The culture was centrifuged (3000 g for 30 minutes) and the resulting pellet was resuspended in 12 ml of lysis buffer (300 mM KCl, 50 mM KH2PO4, 5 mM imidiazole, 10 μg/ml lysozyme, pH 8.0). After gentle agitation at 4 °C for 18 hours, the suspension was subjected to sonication. The insoluble debris was removed by an additional centrifugation step (6000 g for 30 minutes) and the soluble fraction was purified by immobilised metal affinity chromatography (IMAC) on the Profinia Protein Purification System (BioRad, Hercules, California). The protein was eluted in a single 4 ml fraction with 250 mM imidiazole. The amount of TdT generated was calculated from the E280 value and the conversion factor (1 E280 = 0.83 mg/ml). The purity of the enzyme was assessed by SDS PAGE and analysis on the 2100 Bioanalyzer (Agilent, Foster City, CA, USA).
The activity of the purified recombinant TdT was assessed in a TUNEL assay following the protocol of the TUNEL Assay Kit (Roche, Paolo Alto, California). Briefly, Hela cells were either treated with a water control or the apoptosis inducing agent camptothecin. After treatment, 2 x 107 cells were washed in 1 ml of PBS and 100 μl (containing 2 x 106 cells) aliquots were transferred to a v-bottomed 96 well microplate. To each well was added 100 μl of fixation buffer containing PFA. After thorough resuspension of the cells, the plate was incubated for 60 minutes at 20 °C on a shaker. The cells were then pelleted by centrifugation and the fixation buffer was removed by suction. Each cell pellet was washed with 200 μl of PBS, the resuspended cells were repelleted by centrifugation and the buffer was removed by suction. To each well was added 100 μl of permeabilisation solution containing the stain TO-PRO-3. After washing x2 with PBS each cell pellet was resuspended in 50 μl TUNEL reaction mixture (containing 5 units of TdT) and incubated at 37 °C in the dark for 1 hour. The cells were then washed x3 with PBS and analysed by florescence microscopy. The specific activity of the recombinant enzyme was assessed by performing a series of dilutions and comparing it with commercial enzyme of known specific activity from Roche.
Results
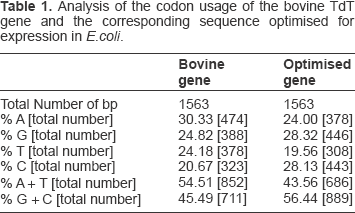
Codon optimisation of the bovine TdT nucleotide sequence resulted in the alteration of 332 codons out of a total of 521, a 64% change. The DNA data for the 2 nucleotide sequences is shown in Table 1. The bovine gene is slightly AT rich in nature whereas the codon optimised gene encoding the same protein is slightly GC rich. It has been reported that AT rich genes are less efficiently expressed in E.coli than neutral or GC rich ones. 14
Analysis of the codon usage of the bovine TdT gene and the corresponding sequence optimised for expression in E.coli.
Small scale inductions, using either 0.5 or 1 mM IPTG, of the different host strains at different temperatures were performed enabling the determination of the optimal conditions to express maximal yields of soluble TdT. It was found that at lower temperatures and at the lower IPTG concentration, higher levels of TdT could be detected in the soluble fraction. Analysis by densitometry showed that approximately 70% of the total TdT was soluble at 22 °C, 60% at 30 °C and 30% at 37 °C (data not shown). A sample of the SDS PAGE analysis of soluble/insoluble samples taken from the 30 °C, 0.5 mM IPTG inductions is shown in Figure 1. Rosetta-gamiB(DE3)pLysS cells induced at 30 °C with 0.5 mM IPTG gave the best yields of soluble recombinant TdT, indicated by the arrow in Figure 1.
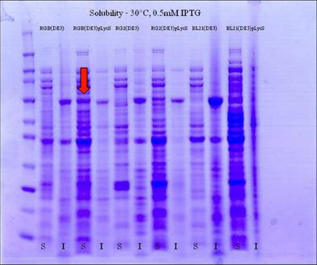
SDS PAGE analysis of the soluble/insoluble fractions generated from 6 strains of E.coli. The arrow indicates soluble TdT from the Rosetta-gamiB(DE3)pLysS strain.
Using the optimisation data generated, a large scale expression was performed. As cell growth at 22 °C was extremely slow, it was decided to express the TdT gene at 30 °C. The material generated was purified by IMAC and its purity was assessed by analysis on the Bioanalyzer (88%). The yield of protein was calculated as described in the materials and methods section; after purification approximately 10 mg of protein was produced from the 500 ml culture.
The biological activity of the recombinant protein was demonstrated by the TUNEL assay. Figure 2 shows Hela cells treated with control and with the apoptosis inducing compound camptothecin. Camptothecin is a natural product from the Camptotheca tree. It is a topoisomerase I inhibitor and induces apoptotic cell death in a variety of different cell types. 15 The permeabilisation reagent containing TO-PRO-3, a fluorescent stain that selectively stains nuclei and not cytoplasmic components, was used to stain the nuclei. 16 The TUNEL reagent containing labeled nucleotides and TdT was used to stain the nicked DNA. Positive staining of nicked DNA generated by the apoptotic activity of camptothecin on Hela cells demonstrated the activity of the recombinant TdT. By merging the images produced by the two stains, the fragmented DNA in the nuclei of the apoptotic cells could be visualized. The activity of the recombinant enzyme made in E.coli was compared to the commercially available enzyme enabling a comparison of specific activity. The specific activity of the recombinant bovine TdT was determined to be 0.5 U/μg of protein.

Fluorescence microscopy of Hela cells treated with either control or camptothecin, then stained with TO-PRO-3 and TUNEL reagent.
Discussion
TdT is a specialized polymerase used extensively in biomedical research. One of its main uses is in the TUNEL assay, an assay that demonstrates the induction of apoptosis. A large number of cancer drugs function by inducing apoptosis in tumor cells, so for this reason the monitoring of apoptosis is a vital component of oncological drug discovery. In recent times there has been a shift towards phenotypic assays as opposed to target-based biochemical screens, allowing biological activity to be assessed at the compound screening phase of drug discovery. Use of the TUNEL assay to screen large libraries of small molecules for anti-proliferative properties is becoming widespread in the pharmaceutical industry. However, this type of high throughput screening is expensive and the price of reagents is not insignificant. For this reason a recombinant source of TdT is required. Heterologous expression of this enzyme in E.coli has been difficult due to the presence of codons in the recombinant gene sequence rarely seen in bacterial gene and there have also been issues of protein solubility and in bacterial expression systems. By manipulating the codon usage of the TdT gene to increase expression and by changing the culture conditions for different strains of E.coli, the generation of active recombinant bovine enzyme in bacteria has been achieved. The method described in this report has yielded 10 mg of recombinant protein per 500 ml culture which translates to 4 x 103 U which would cost over four thousand US dollars from a well known supplier. To conclude the economical production of one of the most expensive reagents required for this routine but important assay has been achieved.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Abbreviations
deoxynucleotidyl transferase;
rapid amplification of cDNA ends;
terminal deoxynucleotidyl transferase dUTP nick end labeling.
Footnotes
Acknowledgements
This work was supported by the Biomedical Research Council (Agency for Science, Technology and Research), Singapore.