
Abstract
Diabetes and its complications are hyperglycemic toxicity diseases. Many metabolic pathways in this array of diseases become aberrant, which is accompanied with a variety of posttranslational protein modifications that in turn reflect diabetic glucotoxicity. In this review, we summarize some of the most widely studied protein modifications in diabetes and its complications. These modifications include glycation, carbonylation, nitration, cysteine S-nitrosylation, acetylation, sumoylation, ADP-ribosylation, O-GlcNAcylation, and succination. All these posttranslational modifications can be significantly attributed to oxidative stress and/or carbon stress induced by diabetic redox imbalance that is driven by activation of pathways, such as the polyol pathway and the ADP-ribosylation pathway. Exploring the nature of these modifications should facilitate our understanding of the pathological mechanisms of diabetes and its associated complications.
Keywords
Introduction
Glucose is a fundamental molecule for life, and its combustion is exploited in all ways to sustain life. While glucose is essential for cellular survival, too much of it is detrimental.1–3 This is the case in diabetes that either originates from or manifests the dysregulation of glucose metabolism. 4 In type 1 diabetes, pancreatic µ-cells are destroyed by autoimmune response, and hence no insulin would be available for stimulating glucose metabolism, leading to diabetic hyperglycemia.4–6 In type 2 diabetes, insulin resistance usually precedes µ-cell dysfunction via a failure of compensation mechanism.7–9 Initially, insulin resistance would aggravate more insulin secretion by increasing µ-cell mass.1,8,10–12 However, such an increase has a limit and will eventually fail to meet the needs for more insulin secretion.9,13,14 Under this circumstance, µ-cells die, insulin levels decrease, and frank type 2 diabetes mellitus develops and progresses.15–18 Regardless of the types of diabetes, it is the persistent level of hyperglycemia that causes all the metabolic problems manifested by diabetic complications, such as blindness, peripheral neuropathy, and chronic kidney disease.6,19,20 Indeed, all the metabolic problems can be attributed to hyperglycemic glucotoxicity.1,2,21–25
Therefore, how glucotoxicity is attained in diabetes? Protein modifications induced directly or indirectly by hyper-glycemia manifest glucotoxicity. In this review, we attempt to summarize a variety of protein modifications in diabetes. We believe that many of these protein modification processes could serve as therapeutic targets or have therapeutic values. We focus on diabetic protein modifications, including glycation, carbonylation, nitration, nitrosylation, acetylation, ADP-ribosylation, and succination. But before expanding on these modifications, we would like to briefly overview the dysregulated glucose metabolic pathways in diabetes.
Glucose Metabolism and Redox Imbalance in Diabetes
When blood glucose level is persistently high, the body will attempt to mobilize all the possible pathways involved in glucose clearance. One such significant pathway is the polyol pathway.26–29 This pathway is usually dormant in nondia-betic state but can be activated to metabolize up to 30% of the glucose pool in diabetes. 30 The pathway involves two reactions, catalyzed by aldose reductase and sorbitol dehydroge-nase, respectively. As shown in Figure 1A, the pathway makes excess NADH by consuming NADPH, hence breaking the redox balance between NADH and NAD+. As the aldose reductase reaction is rate limiting, inhibition of aldose reductase has been shown to prevent the occurrence of diabetes and diabetic complications.31–34 Additionally, glucose is converted into fructose, a sugar molecule whose metabolism bypasses glucokinase and phosphorfructokinase-1 in the glycolytic pathway and thus is less regulated,35–37 thereby inducing metabolic stress. 35 Excess NADH can overload the mitochondrial electron transport chain and drive overproduction of reactive oxygen species (ROS), which can attack proteins and induce protein modifications.35,38 Additionally, consumption of NADPH by the polyol pathway can impair the function of glutathione reductase that uses NADPH to regenerate the reduced form of glutathione (GSH) from the oxidized form of glutathione (GSSG), 39 thus further aggravating cellular redox imbalance. 40
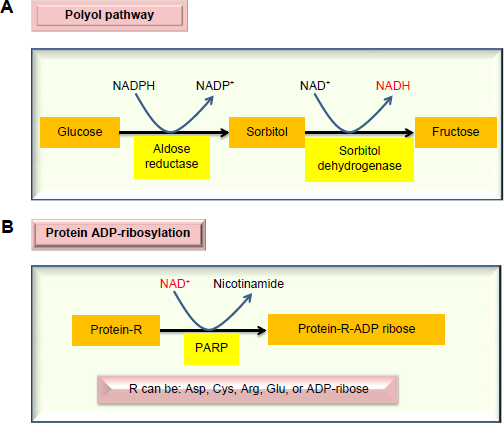
Major enzymatic pathways activated by diabetic hyperglycemia that can impair cellular redox imbalance between NADH and NAD+. The polyol pathway (
Also in diabetes, chronic production of ROS can cause DNA damage.41–44 This damage will activate poly-ADP-ribose polymerase that is evolved to repair the damaged DNA molecules.45–47 As poly-ADP-ribose polymerase uses NAD+ as its substrate (Fig. 1B) and is often overactivated, 48 its activation usually can deplete NAD+ and leads to the further accentuation of redox imbalance, thereby, causing cell death.49–52 It should be pointed out that while activation of both the polyol pathway and the ADP-ribosylation pathway by diabetic hyperglycemia initially appears to be defensive and adaptive, the eventual consequences are lethal. Therefore, diabetes and its complications could be considered as a failure of compensation diseases.53–55
Moreover, diabetic hyperglycemia can also activate other metabolic or signaling pathways. These are summarized in Figure 2, which, in addition to the polyol pathway27,56 and the ADP-ribosylation pathway mentioned earlier, also include the glycation pathway,57,58 the hexosamine pathway,59,60 and the PKC activation pathway.61,62 All these aberrant pathways have been shown to eventually elevate cellular ROS levels,63,64 hence further aggravating cellular redox imbalance and oxidative stress. 38 This redox imbalance is probably the driving force for diabetic ROS production and oxidative stress, which are involved in a variety of protein posttranslational modifications. 63
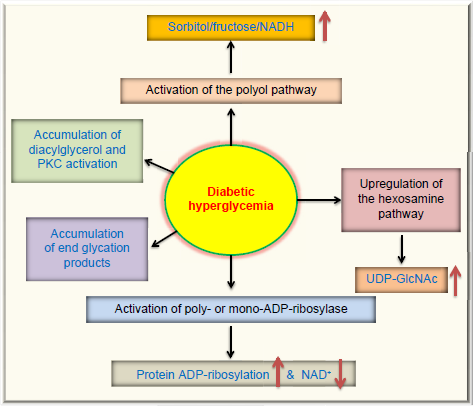
Pathways that are activated or upregulated by diabetic hyperglycemia. In addition to the two pathways shown in Figure 1, diabetic hyperglycemia can also cause activation of the protein kinase C pathway, accumulation of advanced glycation end-products, and upregulation of the hexosamine pathway that fuels the substrate for protein GlcNAcylation. All these pathways have been suggested to be involved in reactive oxygen species production and oxidative stress in the pathogenesis of diabetes and its complications.64,192,193
Protein Modifications in Diabetes
Protein modifications are strategies routinely used by cells to expand their function65–67 but can also reflect the status quo of struggled cellular functions under stressed conditions.68–71 Figure 3 summarizes the types of posttranslational protein modifications in diabetes that are covered in this review. The modifications can be classified into two categories: irreversible and reversible. Irreversible protein modifications include carbonylation, nitration, and glycation, and reversible protein modifications include nitrosylation, acetylation, sumoylation, O-GlcNAcylation, ADP-ribosylation, and succination.
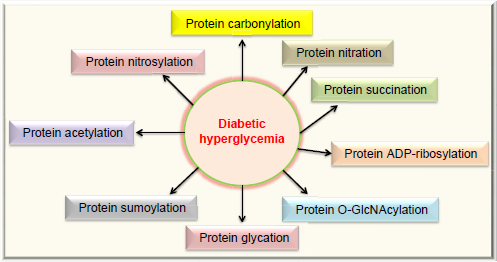
Protein modifications in diabetes reviewed in this article. These modifications include carbonylation, nitration, succination, ADP-ribosylation, O-GlcNAcylation, glycation (AGEs), sumoylation, acetylation, and nitrosylation. All these protein modifications can manifest glucotoxicity in diabetes.
Advanced Glycation end Products
Glucose, in its reduced form, can directly react with proteins.72,73 The reaction usually takes place between the glucose's aldehyde group and the side chain of lysine residues as well as the N-terminal amino groups for given proteins.72,73 The initial species is a Schiff base that can rearrange to form an Amadori intermediate. This intermediate can further rearrange to form varying forms of advanced glycation end products (AGEs).72,73
Glucose can also undergo autoxidation to form ketoal-dehyde and hydrogen peroxide in the presence of transition metals.58,74,75 The resulting ketoaldehyde can further react with the amino groups in proteins. This is followed by the formation of ketoimine via Schiff's base. The ketoimine is then involved in the formation of protein-linked AGEs.58,75,76 It should be noted that fructose can also induce protein glycation. 77
Protein can also be modified by methylglyoxal (MGO), a reactive product in the glycolytic pathway.78–81 MGO is a carbonyl-containing compound and mainly reacts with lysine, arginine, and cysteine residues.79,80,82 The eventual protein adducts are a variety of AGEs that could be structurally distinct.83,84 It has also been reported that MGO can have profound detrimental effects in diabetes.79,85 For example, MGO can impair mitochondrial function in diabetes via modifications of a variety of mitochondrial proteins. 83
Protein Carbonylation
Protein carbonylation is an irreversible process.39,86 Protein carbonyls can be formed directly by ROS attack or indirectly by conjugating to lipid peroxidation byproducts, such as hydroxynonenal.20,87,88 Protein carbonylation can be formed on a variety of amino acid residues’ including histidine’ cysteine’ lysine’ arginine’ proline’ and threonine. 87 Protein carbonyls not only have been used as a biomarker for protein oxidation in aging and disease 89 but have also been shown to impair protein structure and function.90,91 In diabetes, it has been shown that protein carbonylation is increased in red blood cell membranes in diabetic retinopathy. 92 It has also been reported that more plasma proteins show elevated protein carbonyl content in type 2 diabetes. 93 In our own studies’ we have shown that mitochondrial complex I isolated from diabetic kidneys exhibited selective protein carbonylation via the conjugation with lipid peroxidation product hydroxynonenal that contains a carbonyl group. 20 As protein carbonyls are toxic protein adducts impairing protein function and carbonylation can occur to proteins involved in insulin signaling, 94 insulin signaling pathways can be disrupted. 94 Indeed’ protein carbonylation has been suggested to be implicated in insulin resistance,94–96 which is an early event in the development of type 2 diabetes.97–99
Protein Nitration
Protein nitration is also an irreversible protein modification. It occurs on protein tyrosine residues due to attack by peroxynitrite.100,101 As peroxynitrite is formed by reaction between superoxide and nitric oxide,102,103 this modification is related to both ROS and reactive nitrogen species. Glucose is known to be implicated in the formation of nitrotyrosine.104,105 Elegant studies by Koeck et al104,105 have demonstrated that glucose can mediate tyrosine nitration in both adipocytes and β-cells, suggesting a role of glucose-modulated nitration in obesity, insulin resistance, and β-cell dysfunction. Importantly, in both adipocytes and β-cells, specific proteins that underwent nitration have been identified; many of them are involved in glucose metabolism and bioenergetics.104,105
O-GlcNAcylation
This posttranslational modification is a reversible modification occurring on serine or threonine residues. 106 The substrate for this modification is uridine diphospho-N-acetylglucosamine, the end product of the hexosamine pathway.59,60,107,108 As glucose level becomes higher in diabetes, more glucose is fluxed into the hexosamine pathway, resulting in elevated levels of uri-dine diphospho-N-acetylglucosamine that can attach to proteins.107,109 Protein O-GlcNAcylation has been found to be involved in numerous biological processes, such as transcription, redox signaling, apoptosis, autophagy, and protein degradation.110–112 Many proteins involved in insulin signaling can undergo this modification. Moreover, O-GlcNAcylation can worsen glucotoxicity in the liver. For example, O-GlcNAcylation of FoxO1 in hepatocytes can increase its transcriptional activity that then upregu-lates the expression of glucose 6-phosphotase, leading to hyperglycemia by increasing hepatic glucose production. 113 Therefore, protein O-GlcNAcylation has been regarded as a major factor in the development of insulin resistance and diabetes and diabetic complications.109,114,115
Protein S-nitrosylation
This modification occurs on cysteine residues and is also a reversible modification. 71 As cysteine oxidation status can reflect cellular redox status, this modification is tightly linked to oxidative stress and glutathione content. 65 As the modification is reversible, it can regulate protein function either beneficially or detrimentally.65,116 In fact, many studies are now being conducted to explore the beneficial role of this modification in aging and disease.117–121 Nonetheless, S-nitrosylation can play a deleterious role in diabetes. 122 For example, it has been reported that in the early phase of diabetes, the level of protein S-nitrosylation is increased that might lead to mitochondrial dysfunction. 123 It has also been reported that S-nitrosylation is involved in insulin resistance via the modification and inactivation of protein kinase B. 124 It should be mentioned that, similar to this modification, other types of cysteine modifications, such as S-glutathionylation, have also been shown to be involved in the pathogenesis of diabetes and its complications.125,126 For example, hemoglobin shows increased levels of glutathionylation in type 2 diabetes. 127
Protein Acetylation
Protein acetylation is the attachment of an acetyl group onto a lysine side chain in a target protein, and the acetyl group usually comes from acetyl-CoA,128,129 which is a central molecule in metabolism. As shown in Figure 4, acetyl-CoA can be derived from combustion of glucose, fatty acids, alcohol, and amino acids. Under normal condition, acetyl-CoA is channeled into the Krebs cycle for ATP production and is also used for the synthesis of cholesterol and fatty acids. Excess acetyl-CoA usually leads to ketone body production130–132 and nonenzymatic protein acetylation. 133 This modification often occurs on lysine residues134,135 and has been referred to as carbon stress.108,136–138 Except histone acetylation and enzyme-catalyzed acetylation that are well-regulated processes, 139 protein acetylation occurring in cytosol and mitochondria has been widely considered as a pure chemical, nonenzymatic reaction,128,129,133,140,141 although the removal of the lysine-conjugated acetyl groups requires deacetylating enzymes, such as sirtuins.142–144 When the glucose level is high, so is acetyl-CoA that is used as the substrate of acetylation. Hence, proteins can be highly acetylated under hyperglycemic or overnutritional conditions.145,146 When cells switch to use fatty acids as their major energy source, such as under the condition of insulin resistance, whereby glucose cannot enter the cells, 147 the levels of acetyl-CoA can increase dramatically (Fig. 5) and protein acetylation can concomitantly increase. 146 Thus, it has been reported that increased fatty acid oxidation leads to elevation in protein acetylation in the diabetic heart. 148 Additionally, over consumption of alcohol that fuels the production of acetyl-CoA can also elevate protein acetylation.133,149 It should be noted that removal of the acetyl group by enzymes, such as sirtuins, requires the presence of NAD+, which is used as the substrate for deacetylases.150,151 Therefore, a lower level of NAD+ would inhibit protein deacetylation and increase protein acetylation. 152 Hence, protein acetylation is a modification that is highly governed by the availability of fuels and NAD+, the latter being tightly linked to cellular redox balance.153–155 In this regard, it is no surprising that aldose reductase can increase protein acetylation via diminishing the NAD+ levels. 156
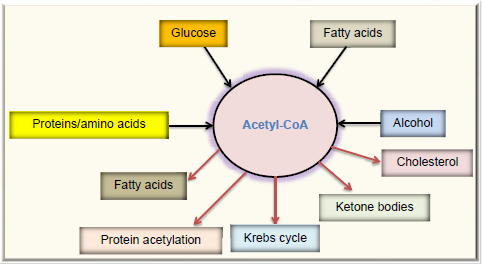
Fates of acetyl-CoA, a central molecule in fuel metabolism. Acetyl-CoA can be derived by combustion of glucose, fatty acids, proteins or amino acids, and alcohol. In normal condition, acetyl-CoA is mainly channeled into the Krebs cycle for energy production. In overnutrition state, acetyl-CoA can be used to store excess energy by forming fatty acids. Acetyl-CoA is also the source for cholesterol synthesis. In starved state, acetyl-CoA is converted into ketone bodies. Acetyl-CoA is also the substrate used for protein acetylation.
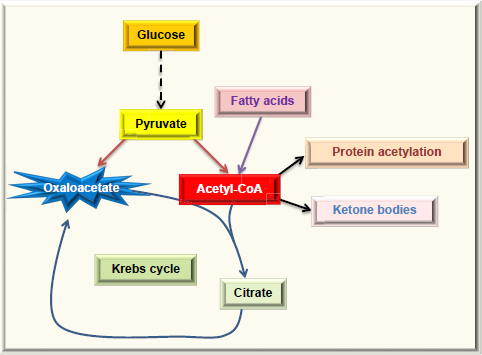
Excess acetyl-CoA drives nonenzymatic protein acetylation. For noninsulin-dependent cells, diabetic hyperglycemia can overload them with glucose, causing the oversupply of acetyl-CoA. For insulin-dependent tissues in diabetes, the cell cannot get enough glucose and will have to use fatty acids as the source of energy. Because oxaloacetate cannot be continuously formed due to lack of glucose, the level of acetyl-CoA could be extremely high, leading to ketone body production and protein acetylation.
Protein Succination
This modification, along with protein acetylation, has also been categorized under carbon stress. 138 Protein succination is due to a conjugation reaction between fumerate and proteins and often occurs on protein cysteine residues.157,158 Any fuel source that would elevate the level of fumerate, an intermediate in the Krebs cycle, would theoretically facilitate protein succination.158–161 Similar to S-nitrosylation, protein succination has been shown to increase in diabetes and its complications.161,162 Protein succination can also impair protein function and cellular redox signaling.38,65,71,163–167 Indeed, it has been reported that protein succination is a manifestation of glucotoxicity in both the glycolytic pathway and the mitochondrial bioenergetics pathway.158,161
Protein Sumoylation
This posttranslational modification refers to the attachment of a small protein, called small ubiquitin-like modifier (SUMO) protein.168,169 SUMOs are covalently attached onto target proteins and can also be detached.170,171 Hence, sumoylation is also a reversible process. Protein sumoylation is known to be involved in protein translocation, protein stabilization, inflammation, redox imbalance, and oxidative stress. 172 In diabetes, SUMO-4 has been implicated in the development of diabetes. 173 The target proteins of SUMO-4 include IKBα, STAT, AP-1, and heat shock transcription factors. 174 Moreover, SUMO-4 seems to restrict its action in pancreas and immune systems as well as in kid-neys.175,176 With respect to regulation of blood glucose levels, sumoylation is known to occur in Glut4, thereby facilitating its translocation onto cell membranes. 177 Sumoylation is also known to occur in protein-tyrosine phosphatase 1B (PTP1B), whereby PTP1B function is inhibited. 178 As PTP1B is involved in a negative regulation of insulin receptor, PTP1B sumoylation is considered to positively regulate insulin signaling. 169 While some of these studies indicate the beneficial role of protein sumoylation, the modification has also been shown to be involved in diabetic pathogenesis.176,179,180 For example, high glucose has been shown to induce sumoylation of Smad4 in mesangial cells, a process likely involved in renal fibrosis in diabetic kidney. 181 Additionally, protein sumoylation has been linked to increased endothelial inflammation, a process known to occur in diabetes and its complications. 182
Protein ADP-ribosylation
This posttranslational modification occurring in several amino acid residues, such as cysteine, arginine, and asparagine, is the transfer of the ADP-ribose moiety of the NAD+ molecule onto a target protein,183,184 and either mono-ADP-ribosylation or poly-ADP-ribosylation can occur. 185 Because NAD+ is used as a substrate for ADP-ribosylases, the process is also highly dependent on NAD+ availability, and activation of ADP-ribosylases can actually deplete NAD+.41–44 This is indeed the case in diabetes as over-activation of poly-ADP-ribosylases and NAD+ depletion have been observed.41,186,187 Accordingly, inhibition of poly-ADP-ribosylases has been demonstrated to prevent the development of diabetes and its complications.188–190 As ROS-induced DNA damage can activate poly-ADP-ribosylases, ADP-ribosylation is thought to be deeply involved in oxidative stress and glucotoxicity. 191
Conclusion
In this review, we have summarized the evidence that post-translational protein modifications can manifest glucotoxicity in diabetes. We have discussed the types of protein modifications that have been, and are still being, intensively investigated in the field of diabetes research. These modifications, including carbonylation, nitration, glycation, O-GlcNAcylation, nitro-sylation, succination, acetylation, and ADP-ribosylation, can affect or modulate the function of the modified proteins’ with consequences that are more often detrimental than beneficial. Importantly, the driving force behind all these modifications is dysregulation of glucose metabolism in diabetes that results in persistent hyperglycemia. Further studies on these protein modifications in diabetes will continue to help our understanding of the pathogenic mechanisms of diabetes and its complications.
Author Contributions
Conceived the idea and wrote the first draft of the article: L-JY. Contributed to the preparation and design of the article and reviewed and approved the final form of the article: HZ, JW, ZJ, L-JY. All authors reviewed and approved of the final manuscript.