
Abstract
The past twenty years have witnessed an explosion of biological data in diverse database formats governed by heterogeneous infrastructures. Not only are semantics (attribute terms) different in meaning across databases, but their organization varies widely. Ontologies are a concept imported from computing science to describe different conceptual frameworks that guide the collection, organization and publication of biological data. An ontology is similar to a paradigm but has very strict implications for formatting and meaning in a computational context. The use of ontologies is a means of communicating and resolving semantic and organizational differences between biological databases in order to enhance their integration. The purpose of interoperability (or sharing between divergent storage and semantic protocols) is to allow scientists from around the world to share and communicate with each other. This paper describes the rapid accumulation of biological data, its various organizational structures, and the role that ontologies play in interoperability.
Introduction
During the 1960s, there was a simultaneous evolution of digital protein and taxonomic inventories. By the 1980s, these had matured and were institutionalized with an attendant proliferation of biological data. These datasets were, however, maintained in closely-guarded proprietary repositories or ‘silos’ with little or no communication between them (Bisby, 2000; Boguski and McIntosh, 2003; Pennisi, 2000). The 1990s were marked by a shift in emphasis from accumulating vast volumes of data to reducing overlap between databases and making use of extant data across various repository locations (Boguski and McIntosh, 2003). This process of increasing communication between databases is known as interoperability—the focus of which is to enable data sharing and comparison.
As the cumulative body of biological knowledge increases, generating a comprehensive and consistent account of biology hinges upon the ability of scientists to draw upon and synthesize vast datasets across distributed digital resources. The ultimate objective of biodiversity informatics is to generate a “global inventory of [all] life on Earth” (Blackmore, 2002, p 365), and is premised on the seamless digital accumulation of distributed taxonomies. Because contemporary biological—particularly ‘omics’ and model organism—databases stress data at the molecular scale, they do not adequately represent the physiology they describe (Boguski and McIntosh, 2003). There is thus a need to compile the cellular features of those organisms into discernible representations of those organisms themselves.
The rise of ‘omics’ science—genomics, proteomics, and metabolomics for the identification and prediction of genetic product components, signatures, and processes (Sauer et al. 2007, p 550)—has contributed the molecular-level information upon which a systems view of biology is predicated. Certainly the complexity of biology resides at the level of gene products (Sauer et al. 2007). In this way biodiversity can be understood as the compendium of the biology of organisms (Chicurel, 2002b). In a computational environment, biodiversity is the hereditary information encapsulated within genetic products and identified via the collective of mappings of several model organism genomes. While the maturation of ‘omics’ has been facilitated in large part by the capacity to seamlessly make divergent data sources interoperable, it has presented a new set of engineering challenges. These include the need to integrate diverse and remote data sources as well as to extract knowledge from digital information post-integration (Thomas and Ganji, 2006).
The paradigm shift the ‘omics’ revolution has created within biology is best exemplified by gene prediction (also known as gene finding), and functional prediction tasks. New technologies such as micro arrays generate huge and ever-changing volumes of data (Buetow, 2005). The rapid growth of genome mapping necessitates the ability to automate gene-calling, or the identification of the individual genes of a genome. Gene finding involves algorithms for the identification of biologically functional regions—or exons—of sequences which explicitly code for proteins (Saeys et al. 2007). These are referred to as
Unlike systems architectures, the integration of which constitutes an ‘IT problem’ (Searls, 2005), data are not semantically transparent. Although a structural linkage can now be easily defined between data sources such that a user can retrieve data on the basis of standardized queries across data sources with conflicting database schemas, this does not render the results of those queries meaningful. A prime example is the notion of ‘gene’—the primitive of modern biology. While the concept of ‘gene’ is still evolving, two dominant concepts exist: the Human Genome Database defines a gene as a DNA fragment that can be interpreted as (analogous to) a protein; whereas GenBank and the Genome Sequence Database (GSDB) consider a gene to be a “‘region of biological interest with a name and that carries a genetic trait’” (Schulze-Kremer, 2002, p 180). Two databases can be developed based on different understandings of ‘gene.’ As a result, retrieving data from semantically orthogonal databases on the basis of a ‘gene’ keyword search can initiate error propagation—in this case in the form of false analogues—in the analysis and subsequent results (Searls, 2005). The complexity of biological terms exacerbates this problem. Even where two variables in disparate databases are semantically equivalent, their relations to other knowledge objects in the data repository may not be. This is referred to as schematic incompatibility and refers to the relative position of the term in a taxonomic hierarchy.
In order to accommodate both semantic and schematic differences between biological databases, ‘omics’ research requires a method of expressing the
The power of ontologies lies in their capacity to provide context for biological semantics. This paper presents the molecular biologist—rather than the computing scientist—with a detailed, comprehensive review of ontologies in biology. We begin with a definition of formal ontology in order to clarify the role that ontologies play with respect to interoperability (or the exchange of data). We describe ontological concepts—and their role bioinformatics—using the examples of two pre-eminent ontological efforts in biology: the Gene Ontology (GO), which is itself part of the umbrella Open Biomedical Ontologies (OBO) initiative. Subsequently we explain how ontologies can be exploited to facilitate information sharing and data integration efforts for bioinformatics with reference to real-world, large-scale biological information portals, namely the cancer Biomedical Informatics Grid (caBIG), and WikiProteins, a proprietary knowledge commons for proteins.
Furthermore, we describe a methodology for using ontologies as a basis for comparing semantics across health registries in order to illustrate how medical informaticians have imposed interoperability on disjunct datasets. Once semantic and schematic heterogeneity is resolved between datasets, we explain how ontologies can be used to facilitate knowledge creation tasks in biology, such as automating gene/protein annotation and functional prediction.
To provide a global overview of ontologies for biology, we also draw upon a related community of research—health/medical informatics—which uses and shares with bioinformatics a series of knowledge representation constructs for the capture of biological information. Both use genetic information in the era of “‘post-genome’ science” (Boguski and McIntosh, 2003, p 233). For instance, knowledge sharing protocols developed in the field of health informatics are shared by bioinformatics researchers for resolving semantic heterogeneity in databases. Wang et al. (2005) for example use Protégé—an open-source ontology editing and knowledge acquisition software authored by Stanford Medical Informatics (2005)—as the knowledge representation platform for their mediation architecture. There are similarly links between bioinformatics and biodiversity communities. The tools of bioinformatics—many of which emerge from health informatics—are ideally suited to the objectives of biodiversity research, particularly conservation science (Sugden and Pennisi, 2000). This paper nevertheless emphasizes bioinformatics.
Ontologies
In philosophy, ontology has traditionally been understood to be the essence of being—or what something really is (Schuurman, 2006). In the information sciences, an ontology is a fixed universe of discourse in which each element (e.g. field name or column in a database) is precisely defined (Gruber, 1993). In addition, each possible relationship between data elements is parametized or constrained. For example, DNA may comprise chromosomes but not the reverse. In an ontology, these relationships are made explicit formally.
The prefix ‘formal’ refers to the property of machine-readability (Agarwal, 2005). In other words, a
Scientific or systems ontologies contain three levels of formalization. The first is the conceptual, which is then translated into a formal model of the data elements in the ontology (e.g. proteins) and the possible relationships between them. The final stage or level is the development of code that can be run by computers (Schuurman, 2006). Ontologies are structured much like a biological taxonomy with general concepts appearing at the top of the tree and becoming more general as one traverses down. The hierarchical schema, however, is only a ‘shell’ that can accommodate the concepts and their relations particular to a domain (Rector, 1999; Schulze-Kremer, 2002). It must be populated by domain knowledge expressed in a formal semantics—a computing syntax such as a markup language—that allows all entities declared into the ontology to be precisely defined and their interrelationships given strict parameters with the goal of enabling realistic biological models.
Formal semantics permit the distinction of concepts declared into the model (Sowa, 2000). To satisfy the strict criteria of formal ontology building, the formal semantics used to instantiate an ontology should be premised on a formal logics particular to some logical algebra (Smith et al. 2005)—such as description logics (DL)—which contain predetermined rules for “when two concepts are the same, when one is a kind another, or how they differ” (Rector, 1999, p 239–52, p. 10). These rules must furthermore be expressed in some machine-readable syntax—in this case, a knowledge representation language such as the Web Ontology Language (OWL). Such rules govern the expression and processing of

The formalization process. Moving from a concept of a particular gene to its encoded reification and ontological representation. Note how the entity (fruit fly) becomes increasingly represented in digital database format as it is formalized, or abstracted from its real-world form. The entity loses dimensionality, while researchers gain the advantage of computational function. Figure 3 illustrates in more detail the role that entity descriptions—or annotations—play in creating a larger standardized digital knowledge environment for bioinformatics. Note that any gene product many have more than one annotation in the same branch (see Molecular Function this example), and can be annotated in three different branches of GO (Cellular Component, Biological Process, and Molecular Function) (FlyBase 2007, The Gene Ontology Consortium 2077).
The ability to define relationships between concepts distinguishes formal ontologies from earlier integration and interoperability approaches. How they are expressed are detailed in the subsequent sections on the GO and OBO efforts. Relationships are an expression of the
Formal ontological expressions are stated as propositional triplets consisting of
In a strict definition of formal ontology, the axiomatic logics serve to underwrite a formal notation for content specification. For example, DL comprise the logical
This structure moreover makes the ontological model amenable to implementation in a software environment (Smith et al. 2003) in order to allow for the kind of intelligence described using the example of a cardinality restriction in the instance, ‘a man must have at least one tests’. The taxonomic structure of formal ontologies captured using logical notation and expressed in a knowledge representation language allows the semantics of concepts to be computed on the basis of concept inheritance. This is known as
Ontologies—with their hierarchical structures—capture the semantic granularity of biological databases. The property of inheritance allows the computer to process, for example, that the concepts used to annotate two respective sequences are both ‘children’ of the same meta-concepts (i.e. they are a kind or part of a the same overarching concept; alternatively, members of the came class) (Lord et al. 2003). This permits researchers to locate regions of exact correspondence as well as those with a high degree of similarity. Entities may relate but are not synonymous—for example, where ‘protein’ is a subclass of another concept, ‘gene products’ (Ahlqvist, 2004; Ahlqvist, 2005). This does not dictate that proteins and genetic products are one and the same, but rather allows the expression of a membership relation at a much finer semantic resolution such that proteins can be understood as one, but not the sole, kind of gene product (which also includes RNA).
Thus far, we have described the problem of semantic and schematic heterogeneity and introduced ontologies as a means of mitigating the problem. The formal implementation of ontologies—as well as necessary conditions for formality—has also been discussed as well as its advantages for promoting computer reasoning. In the next section, we describe in detail the genesis and development of a bioinformatics portals, GO and its role in biological data interoperability. In addition, we briefly illustrate the implementation of ontologies in two database as well as an ontology-based method for comparing data from different registries or jurisdictions.
GO: Ontology in Practice
The use of ontologies for bioinformatics is being driven by the proliferation of genome-scale datasets and the diffusion of the Internet and its protocols for data sharing and exchange (Blake, 2004). Bio-ontologies fulfill two central functions for the biological domain—first, they “clarify scientific discussions” by providing the vocabulary and terms under—and with which—such discussions take place, and second, they enable data discovery across distributed data resources (Blake, 2004, p 773). The pre-eminent bio-ontology is the (GO), a Web-based, open source knowledge resource for bioinformatics and the second-most cited biological data resource after UniProt (Galperin, 2006).
The GO project evolved as a joint endeavor between three model organism databases: FlyBase, Mouse Genome Informatics Database (MGI), and the
The chief impediment to this task were not the unique identifiers for the gene products themselves as researchers had been tapping into protein and gene databases such as GenBank and Swiss-Prot, TrEMBL and PIR for decades (the latter three joined to form the Universal Protein or UniProt protein repository in 2002). Because sequences are unique, they could be easily accessed on the basis of sequence characteristics (though there was sequence redundancy between protein repositories). Computationally, because sequences can be quantified, this is a trivial integration task that simply requires the normalization of unique codes (Schuurman et al. 2006). Rather, it was the functional descriptions of gene products that proved challenging. Integration had to proceed within the context of the molecular and biological characteristics of each gene product identified (Lewis, 2005). In an attempt to solve the problem, informatics experts from the three original participating model organism databases devised functional classification systems in the hopes that these precursors to the GO would facilitate interoperability. What soon became apparent, however, was that these functional classifications were not common between organisms (Lewis, 2005).
In other words, the annotation was not consistent from one database to the next. Gene annotation is defined as the “task of adding layers of analysis and interpretation to … raw sequences” (2002, p 755). This includes information about their function, position relative to coding/non-coding boundaries, participating process, etc. (Chicurel, 2002b). Annotations constitute a set of
The GO Consortium formed as a response to the pervasive semantic heterogeneity of biomedical data and its lack of formality. Indeed the GO was designed for making historically free-text based annotations tractable (Lord et al. 2003). The three participating database programs agreed to work in concert to provide the biological community with a consensus-driven framework to guide the annotation of gene products such that their structure (e.g. how molecular function is described and which part of the description occurs in what syntactic order) and semantics (the terms and concepts) are consistent. The result was the GO—a “structured, precisely defined, common, controlled vocabulary for describing the roles of genes and gene products in any organism” (Ashburner et al. 2000, p 26). The GO is not a taxonomy or index of all known proteins and gene products, but rather provides a standardized set of names for genes and proteins and the terms for characterizing—or ‘annotating’—their behaviors (Gene Ontology Consortium 2007).
Gene product semantics are organized into three categories which capture the primary ‘aspects’ of genes: i) biological process, which captures the larger process in which the gene product is active; ii) molecular function, the biochemical function a gene product contributes to that process, and iii) cellular component, the location in the cell where that particular function is fulfilled or expressed (Ashburner et al. 2000; Gene Ontology Consortium 2007; Hill et al. 2002; Lord et al. 2003). Concepts or terms constitute nodes, and vectors referred to as
Each of these three separate annotation categories—biological process, molecular function, and cellular component—is represented as its own directed acylic graph, or DAG (Ashburner et al. 2000; Hill et al. 2002; Lord et al. 2003). A DAG is a data structure similar to a tree which represents knowledge hierarchically, mirroring the taxonomic structure of biological knowledge. Any entity can point to any other entity in the mathematical space; this is, however, a direction, and non-recursive, encoding. In other words, concepts can point to other entities in the model, but those entities do not ‘point back’ as in OWL. Indeed the DAG can be considered to be the native knowledge representation (KR) language of the GO (Aranguren et al. 2007). Unlike the KR languages introduced above, however, DAG semantics are not predicated on a formal logic as they are in the case of OWL. Rather, machine readability is instructed by the directional links between pairs of concepts. Semantic ‘edges’ (relationships) in the DAG are simply “ordered pairs of nodes” (Aranguren et al. 2007, p 61). Pointers are like edges in the sense that their semantics are directed, and are labeled with the relationship that associates related classes. These associations are of only two relations:
The DAG is available in many file formats— XML, OWL—but the most common formal notation in which GO ontologies are rendered is the Open Biomedical Ontology (OBO; described in more detail below) flat file structure which is underwritten by a modified subset of Web Ontology Language (OWL) description logics (DL) concepts for content specification (Gene Ontology Consortium 2007).
Like OWL, OBO is an ontology language, and standard ‘file format’ for GO annotations. It is however less expressive than OWL. These relations are unidirectional and linear as per the DAG data model and do not require the recursive relational declarations (where the reciprocal or inverse of a relationship is also encoded) characteristic of OWL statements. Thus a flat file structure that only supports sequential reading is appropriate for the GO because relations are read from broader or general to more specific or precise concepts.
At the level of the database, the GO is represented as a structured vocabulary; more specifically, as gene product annotations expressed using concepts and their tripartite (biological, molecular, and cellular) structure defined in the GO (Hill et al. 2002). The GO is not considered an informatics ontology in the full sense of the term because it has not been designed to be deployed within software environments which execute semantic inference on the basis of logical semantics (Smith et al. 2003). Moreover it does not fulfill the conditions of formality identified by Smith et al. (2005). Rather it is considered and referred to by its engineers as a “controlled vocabulary” (Ashburner et al. 2000, p 26). The nevertheless has many of the characteristics of a formal ontology: machine-readability, formal notation, a hierarchical knowledge structure, and relational associations between concepts. In other words, the GO may be considered a partial implementation that uses many concepts of formal ontology. Part of the reason, however, that the GO is only a partial implementation is that it was designed to be operational within existing infrastructures, requiring no changes to existing architectures.
Notwithstanding, the GO provides the standard vocabulary for semantic integration and automated tasks for bioinformatics. As such it is more than merely a sophisticated data dictionary. Whereas controlled vocabularies or data dictionaries provide a definition of the terms used by a community of practice and these may indeed be machine-readable and thereby formal, a nomenclature does not capture the hierarchical representation of knowledge nor the corresponding relations between all concepts in the data space, and thereby does not support computational reasoning (Schulze-Kremer, 2002). Several terminological systems such as SNOMED (Systematized Nomenclature for Medicine) and MeSH (Medical Subject Headings) have, however, been mapped to the GO (Searls, 2005).
The GO is a
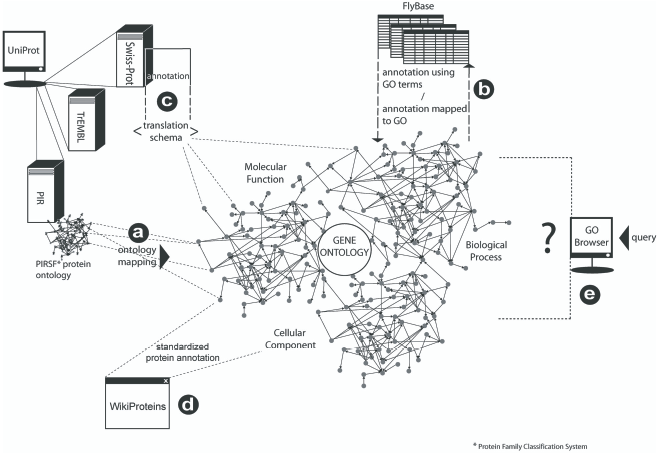
The Gene Ontology as a global ontology for bioinformatics. Smaller scale bioinformatics ontologies almost invariably map to the GO (
A global ontology paradigm is appropriate for the domain because there is a finite, though as yet not fully discovered or known, body of genetic information shared between all life on Earth (Ashburner et al. 2000). Accordingly there is no need to build
The alignment of currently non-compatible ontologies to the GO is one avenue for its
The GO was designed specifically to account for molecular function, biological process, and cellular components of gene products. It lacks the semantics to describe the physical attributes of genes, to describe a protein family, or to account for experimental processes and diagnostic procedures (Wolstencroft et al. 2005). There are both proprietary and open ontologies with richer semantics for more specific description tasks for biology either being developed or presently available (for examples, see Chicurel, 2002; Peters and Sette, 2007; Schulze-Kremer, 2002). The majority, however, are designed with mapping to the GO in mind (Buckingham, 2004b; Chicurel, 2002b).
Open Biomedical Ontologies
GO is thus not the sole ontology for biology. Indeed there is a need for ontologies to parallel the GO programme. The GO is only one—but certainly the most prominent—ontology effort which contributes to the Open Biomedical Ontologies (OBO) initiative (Gene Ontology Consortium 2007). The OBO Foundry is an umbrella for over 60 bio-ontologies (Smith et al. 2007, The Open Biomedical Ontologies 2007). It provides guidelines for ontology development, and indeed ontologies such as GO have been restructured in line with OBO specifications. As indicated above in the detailed discussion of GO, OBO is also its own ontology format (although OBO does provide an extensive suite of translation schema for mapping OBO representations to, for example, OWL) (The Open Biomedical Ontologies 2007). The benefit of this is that, given domain consensus, it provides for uniform representation and thereby increased interoperability. For example, disparate cell-type ontologies including the GO are now integrated into a single ontology that is itself being aligned to a singular implementation. OBO participates in the National Center for Biomedical Ontology and is slated to become a centralized resource of its emergent BioPortal in support of bioinformatics knowledge discovery and sharing.
Ontologies in Support of Bioinformatics
The largest public contributor of annotations to the GO project is the Gene Ontology Annotation Database (GOA) (Camon et al. 2004). While annotation is the central organizing principle and
One of the primary objectives for bioinformatics to realize is the automation of annotating cross-matches between databases (Ashburner et al. 2000). The electronic generation of annotations based on homology is particularly desirable as the manual curation of gene-oriented databases is time consuming and non-trivial for humans (Chicurel, 2002b). The GO facilitates the automatic annotation of gene products at the database level. GOA for instance uses GO terms to generate annotations for the UniProt Knowledgebase (The consortium of SwissProt, TrEMBL, and PIR-PSD protein databases) (Camon et al. 2004). Existing data held in UniProt are electronically associated with or translated into GO terms on the basis of a defined mapping file used to facilitate the conversion of keywords in the constituent databases to tractable GO representations (Camon et al. 2004). Once the semantics are consistent between data sources, biologists who have identified a new sequence, for example, can navigate the GO via an interface known as an
The GO can be used to automate the following services: database annotation, GO extension (automating the transfer of new annotation concepts
Once the protein or gene of interest is isolated, its location confers more information than a binary indication of its absence or presence in a database. Not only do we know about the occurrence of a protein, for example, but we are told something
Ontologies can further be used as a basis for exploring datasets. We have devised a methodology called
The gestational hypertension/hypertension example above would indicate that hypertension experienced during pregnancy is a more general concept which includes gestational hypertension but also encompass pre-existing hypertension. In some databases, hypertension and pregnancy-induced or gestational hypertension are not differentiated from chronic or pre-existing incidences of disease. Alternatively, in other databases, these concepts are distinguished from each other on the basis of the periodicity of disease onset such that chronic hypertension and pregnancy-induced or gestational hypertension are disjoint (database

Ontology mapping. An ontology for hypertension resulting from the merging of hypertension concepts in the British Columbia Reproductive Care Program Perinatal Database Registry (BCRCP PRD) and the Canadian Perinatal Database Minimum Dataset. The resulting output ontology shows the hirerarchical nesting of hypertension semantics originating in respective databases in relation to each other.
Another instantiation of the ontology-based metadata concept similar to our implementation is WikiProteins, a structured semantic space for capturing the context—biological, physiological, chemical, etc.—of proteins and then sharing that collaborative knowledge with other biologists in real-time (Giles, 2007; Wiki For Professionals 2007). Historically, the problem with metadata has been that it is so labor intensive and never updated (Schuurman and Leszczynski, 2006). WikiProteins provides a mechanism for sharing the labor and ongoing maintenance by participants. This collaborative Web-based workspace facilitates the open curation of protein-specific information by providing biologists and bioinformaticians with a means of contributing to the cumulative body of biological knowledge. At the moment, it serves UniProt and GO descriptions for the annotation of proteins via a series of standardized forms or ‘slots’ for their description. This consists of definitions, attribute-value relations (e.g. a protein can be given the attribute “tissue” with the value “[e]xpressed in muscle fibers”), and provisions for disambiguating sequences or instances of proteins by identifying synonyms, disjoint concepts, alternate spellings, etc. (Wiki For Professionals 2007). Curators can link their descriptions or proteins to other citations, references, and publications indexed in PubMed. Moreover, the wiki concept ensures that these annotations are self-validating. Other users can go in and add or revise the annotations. For example, using the “tissue” example above, a subsequent curator can reify this protein as “[e]xpressed in muscle fibers and
As our non-automated method for data discovery and WikiProteins for protein knowledge exchange illustrate, ontologies are not standalone solutions for interoperability but rather comprise a component of or input to large-scale interoperability infrastructures. Indeed ontologies are knowledge representations and
Conclusion
We have described the challenges inherent in semantic integration of biological databases. Many of these are common to all semantic integration realms and are based on the problem of language not being transparent across institutional and user environments. In biology and other disciplines, ontologies or strict paradigmatic taxonomies have been used to mitigate the problems associated with semantic integration. Ontologies are a means of conveying context associated with semantic terms so that their meaning is transparent between multiple data users. This paper has described in depth the use of ontologies for data integration in biology. Biology has led the scientific world in developing a number of unique approaches to semantic integration. These unique ontology-based integration efforts include ontologies such as the Gene Ontology (GO), and frameworks that exploit their machine-readable semantics to support bioinformatics tasks, such as the cancer Biomedical Informatics Grid (caBIG) and the incipient WikiProteins knowledge community. In addition, we introduced the concept and early implementation of ontology-based metadata in order to demonstrate the role that context plays in clarifying hierarchical and schematic relationships between like but non-equivalent semantic terms in different databases. Each of these is a unique approach to surpassing the problems associated with a lack of congruity in language and meaning across scientific databases.