
Abstract
The COVID-19 coronavirus, which primarily affects the lungs, is the source of the disease known as SARS-CoV-2. According to “Smoking and COVID-19: a scoping review,” about 32% of smokers had a severe case of COVID-19 pneumonia at their admission time and 15% of non-smokers had this case of COVID-19 pneumonia. We were able to determine which genes were expressed differently in each group by comparing the expression of gene transcriptomic datasets of COVID-19 patients, smokers, and healthy controls. In all, 37 dysregulated genes are common in COVID-19 patients and smokers, according to our analysis. We have applied all important methods namely protein-protein interaction, hub-protein interaction, drug-protein interaction, tf-gene interaction, and gene-MiRNA interaction of bioinformatics to analyze to understand deeply the connection between both smoking and COVID-19 severity. We have also analyzed Pathways and Gene Ontology where 5 significant signaling pathways were validated with previous literature. Also, we verified 7 hub-proteins, and finally, we validated a total of 7 drugs with the previous study.
Introduction
The COVID-19 coronavirus, as known as the SARS-CoV-2 virus, is an extremely contagious disease that mostly affects the lungs1,2 since 2019 in the month of December. It has been resulted that more than 540.92 million people are newly affected while 6.3 million people losing their life in all over the world. 3 According to a World Health Organization (WHO) investigation, it is estimated that it caused 14.9 million deaths in direct and indirect ways between 2020 and 2021. 4 The virus has spread to nearly every country and has caused widespread social upheaval. 5 As per that, a history of smoking raises the risk of developing COVID-19 with a severe illness and smokers are more likely to experience it.6,7 Furthermore, Chinese COVID-19 patients indicated that 32% of smokers (including current and former smokers) had a severe pattern of COVID-19 lung disease when they were admitted, compared to 15% of those who had never smoked. 8 Another research represented that tobacco smoking affects and damages the epithelial cell of the lungs. 9 Because of that, it facilitates the virus entry into the lungs’ epithelial cells, 10 causing more severe symptoms and increasing the risk of death.8,11 Moreover, the ACE2 (angiotensin-converting enzyme-2) receptor is also highly expressed in this type of patients and smoking helps to express the ACE2 receptor. 12 According to a prior study, the main host cell receptor of COVID-19 is ACE2 receptor which is essential for the virus to enter cells in order to cause infection. 13 In addition, the outcomes of cigarette smoke can be worsen in COVID-19 patients by up-regulating the angiotensin-converting enzyme-2 (ACE-2) receptor that SARS-CoV-2 uses to enter the host cell and trigger a “cytokine storm.”14,15 Patients with COVID-19 had fewer natural killer (NK) cells, which affect adaptive and innate immunity and significantly increase CD8+ T cell expression. 16 Similarly, smokers had a larger percentage of CD8+ T cells and fewer circulating NK cells than nonsmokers.14,17 In another study, smoking affects the platelet directly and accelerates blood coagulation.18,19 Patients with COVID-19 have also been reported to have coagulation abnormalities, primarily thrombotic complications, and the occurrence of venous and arterial thrombotic abnormalities in COVID-19 patients who are admitted to the intensive care unit (ICU) may climb 31%. 20 Smoking on a regular basis has been shown to facilitate the progression of COVID-19 and rise the threat of various complications in COVID-19 patients. Consequently, we have attempted to identify shared common gene between COVID-19 and smoking. Moreover, there is still a requirement for the relationship between COVID-19 and smoking in bioinformatics analysis that is too important for future research. Finally, our study helps us to understand an important advice about the severity of COVID-19 patients with a smoking history and a resource for pharmaceutical companies wishing to create effective drugs that will reduce the number of difficulties for COVID-19 patients who were ex-smokers. We created 9 (Differential Expressed Common Genes [DEGs], Gene Ontology (GO), Pathways, Hub-Protein, Transcription Factor, Gene Interactions, Gene miRNA Interaction, Protein-Drug Interaction, and Protein-Protein Interaction [PPI]) bioinformatics systems in response to these findings to analyze data on gene expression from COVID-19 patients and people who smoke to better understand their association with each other. To understand that deeply, we have analyzed above 9 biological systems between COVID-19 and smoking in this study. We have depicted our methodology of the hypothesis as shown in Figure 1.

Graphical representation of the underlying procedure of this research.
Materials and Methods
Dataset employed in this study
The National Center for Biotechnology Information (NCBI) 21 and the Gene Expression Omnibus (GEO) are the sources of our data sets. 22 We chose the data set based on a number of criteria, including sample size (minimum 8), and only one condition control versus case (case vs treatment condition was avoided), RNAseq data, and datasets without human creatures. The data set chosen was count data, and differential expression analysis was performed correctly. Unfavorable formatting and datasets that were not relevant to our research goals were excluded. At least 2 datasets, particularly relevant to Covid-19 and smoking, were chosen based on the most appropriate criteria. The datasets with the association numbers GSE152418 and GSE76326 were analyzed from RNA-seq human gene expression datasets. Both have control and case (disease-affected) samples, which are both ideal for our study. We assayed the datasets which bear the association numbers GSE152418 23 and GSE76326 24 from RNAseq human gene expression datasets. Both have control and case (disease-affected) samples, individually perfectly suited for this study. The COVID-19 dataset (GSE152418) contained 6 COVID-19 patients and 3 control or healthy people using Illumina NOVAseq 6000 (Homo sapiens). The Smoking dataset (GSE76326) contained an assessment of the immunity of 6 Smoking patients and 3 control or healthy people using Illumina HiSeq 2000 (Homo sapiens) as shown in Table 1.
The summary of the transcriptomic data and analysis.
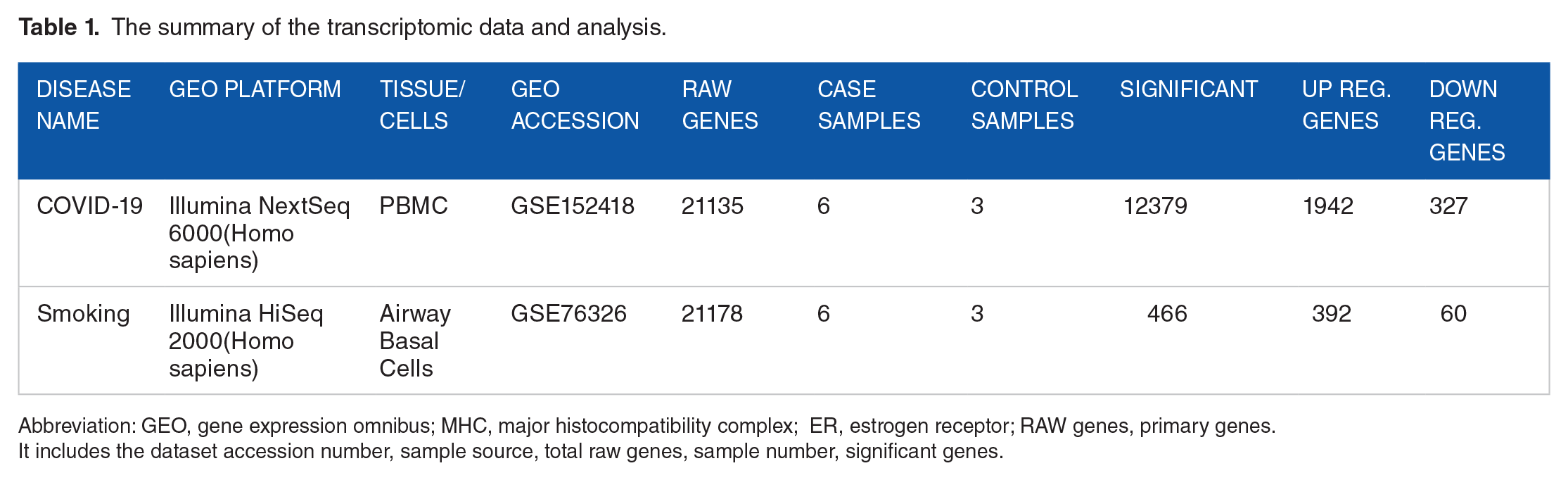
Abbreviation: GEO, gene expression omnibus; MHC, major histocompatibility complex; ER, estrogen receptor; RAW genes, primary genes.
It includes the dataset accession number, sample source, total raw genes, sample number, significant genes.
Preparation and recognition of genes with differential expression
Again from GREIN (it is the most common and unique source of GEO RNA-seq data which is a combination of a huge number of back and front end data and user-friendly interface. 25 Gene Expression Omnibus, we acquired gene regulation RNA-sequence datasets. Using the Rstudio DESeq2, genes with differential expression (DEGs) of COVID-19 as well as smoking-related RNA-Seq count data were found. 26 In order to standardize the results, the geometric mean of each gene was calculated across all samples in DESeq2. Then, DESeq2 automatically filtered out low-expression and outlying genes using Cook’s distance. We have filtered the significant DEGs based on 2 conditions such as adjusted P-value ⩽ .05 and log2F C ⩾ 1. We found common DEGs between COVID-19 and smoking by creating a Venn diagram as shown in Figure 2.

The gene-expression analysis summary was shown in the figure. (A) Venn diagram showing the shared biomarker genes respectively COVID-19 as well as smoking. (B) Bubble plot of common biomarker genes and their corresponding Log2 fold change and adjusted AP-value.
Diseasome network
Diseasome is a representation of disease maps, where the diseases are the nodes and the different molecular connections between the disease-associated cellular components (CCs) are the links. We use InteractiVenn as a bioinformatics computational tool of Diseasome Network for detecting common gene between COVID-19 and smoking. The discovery of such connections between diseases not only aids in our understanding of the molecular relationships between various phenotypes, which are frequently addressed by various medical sub-disciplines but also aids in our understanding of why particular groups of diseases co-occur. 27 The disease map offers a vibrant visual representation that helps in conceptualizing the links between genes and diseases. 28 Diseasomes show local clusters of diseases whose molecular links are well recognized, and they also indicate common pathophysiology between diseases by using pleiotropic genes. 29 A bipartite graph with 2 separate sets of nodes is the best way to depict the disease. The illness nodes in the first set and the disease gene nodes in the second. If a gene’s mutations are linked to a disorder, the disorder and the gene are then linked. A bipartite network is one in which, like in the diseasome, the linkages always interconnect the 2 nodes from 2 distinct groups of nodes. Data from a wide range of sources must be integrated because of the complex relationship between medications, genes, and diseases. Therefore, there is a growing need for new tools to integrate, represent, and visualize heterogeneous biomedical data in order to extract biologically significant information. 30
Gene ontology and molecular pathway identification
In order to identify functional pathways related to specific diseases, many bioinformatic techniques frequently put a heavy emphasis on assessing the significance of a gene set’s similarity to other gene sets with observations. 31 We used EnrichR gene set enrichment analysis to identify the Gene Ontology (GO) terms as well as pathways connected to the overlapped DEGs for both COVID-19 as well as smokers. The analysis emphasizes GOs as well as pathways, which will undoubtedly aid in our quest to fully understand the biological processes (BP) that involve both conditions. 32 The Gene Ontology (GO) project’s primary aim is to develop a standard set of gene dictionaries for describing gene products across all known species. There are 3 categories in GO terminology: CC molecular function (MF) as well as BP. 33 For our study, we only took into account the BP, though. Pathways demonstrate their importance in determining how living things react to stimuli. In the field of life sciences, pathway analysis is usually employed to assist researchers to comprehend the molecular mechanisms that underlie the elevated biological data. It might also explain how various illnesses or conditions are related to one another. 34
Protein-protein interactions analysis and hub-protein
All BPs depend on networks of protein–protein interactions (PPIs), which show the physical contacts, respectively, 2 or more biomolecules. 35 We also used the STRING database 36 as well as Network Analyst 37 to construct PPI networks in between proteins of DEGs, which are based on their physical interactions with one another. Proteins are represented in these networks as nodes, as well as protein interactions, are expressed as edges. Protein-protein interaction analysis with topological characteristics, such as a degree of interaction greater than 15 degrees, was used to identify proteins with strong interactions. Highly interacting proteins are known as Hub Proteins. 38
Transcription factors analysis and miRNA interactions analysis
miRNA is the name of a group of small Biomolecules (21-23 nucleotides) that control the synthesis of proteins but don’t encode any proteins. 39 MicroRNAs (miRNAs) are a type of non-coding small RNA that can regulate gene expression by combining with the 3′-untranslated region (UTR) of a target mRNA. 40 Gene transcription is regulated by TFs, which are modular proteins that bind to the promoter regions of their target genes. As a result, TFs have the potential to increase both the rate of gene expression and the number of proteins made in this way. 39 We investigated the interactions between DEGs and transcription factors (TFs) and DEGs and microRNAs in order to determine the controlling biomolecule (i.e. miRNAs and TFs) that control DEGs of involvement at the transcriptional and post-transcriptional level (miRNAs). With the help of the JASPAR database, the DEGs-TFs interaction was studied 41 Utilizing TarBase 42 and miRTarBase, 43 we used miRNA-DEG interactions. Using Cytoscape’s Network 44 Analyzer and Network Analyst 37 the topological analysis was carried out. The TFs were filtered out of the DEGs-TFs network based on ⩾ 20. The miRNAs were chosen depending on the ⩾ 15 from the DEGs-miRNAs network.
Drug–protein interaction
Studying protein–drug interactions is essential to comprehending the structural requirements for ligand affinity.45,46 The DrugBank 47 dataset is also used to construct protein-drug interactions. We used this (version 1.0) 48 database to find potential medications that significantly interact with genes. The DSigDB is a free online tool that provides a list of drugs and the genes they are meant to target (GSEA). There are currently 22527 gene sets, which include 17389, in this dataset, by using EnrichrR 49 web server and DSigDB database, we chose the enriched drugs for the DEGs utilizing the statistical criterion, adjusted P-value ⩽ .05.
Result
Evaluation of transcriptomic data for gene expression
Both databases were made using prior research that included earlier published GWAS results. To study how smoking and COVID-19 patients’ genes are expressed, we collected RNA-Seq data from GREIN or NCBI. Table 1 shows that the COVID-19 data came from the “Illumina Next Seq 6000 (Homo sapiens)” GEO platform, while the smoking data came from the “Illumina HiSeq 2000 (Homo sapiens)” platform. The RNA-seq data for COVID-19 came from the “Peripheral Blood Mononuclear Cells” study, and the RNA-seq data for smoking came from the “Airway Basal Cells” study. For COVID-19, we’ve chosen the dataset with the GEO accession ID GSE152418 23 and the Smoking dataset with GEO accession ID GSE76326. 24 Nine samples total—6 for the case and 3 for the control—were used in COVID-19. After differential expression analysis, also known as generated signature data, 21135 differentially expressed (DE) genes for COVID-19 were discovered. Then, 2 conditions were taken into account for the signature data, including “Log2 Fold-Change” and “Adjusted P Value.” Following the application, we identified 12379 significant genes by applying the criteria that the adjusted P value be less than .05 and the abs(LogFC) be higher than or equal to 1.0. Of the significant genes, 1942 are up-regulated and 327 are down-regulated.” However, there are a total of 9 samples in the Smoking dataset; 6 of them come from the case group (regular smokers), while the other after producing signature data, we identified 21178 differential expression genes. Then, using the same standards as for smoking, we found 466 significant genes. Of these, 392 are expressed positively (up-regulated), and 60 are expressed negatively (down-regulated). Then, we made a comparison between the COVID-19 upregulated genes and the upregulated smoker genes. We also compared the genes that were down-regulated in both conditions. There are 28 common genes that are upregulated and 9 common genes that are downregulated between COVID-19 and smoking as shown in Figure 3. The most important genes that are shared up-regulated are CHPF, GP1BB, HIST1H4J, EGFL7, PLXNB3, MPL, JSRP1, KLC3, zTOX2, EVA1B, APOE, GAS2L1, NTSR1, CAVIN3, IGFALS, ADAMTS7, PITX1, RNF17, BCAR1, NPW, HGFAC, TCF19, SPDYE2B, GRIN3B, LMNTD2, SLC16A8, CYP2W1, PTGER1. Moreover, the most prominent down-regulated genes shared by COVID-19 and smoking: TIRM27, NFKBIL 1, WDR46, C4A, HLA-DQB1, HLA-C, SKIV2L, HLA-DPB1, CARS.

Up-regulated and Down-regulated genes between COVID-19 patients and smokers are represented separately. (A) Up-reg. (B) Down-reg.
Analyzing the functional associations between pathways and gene ontologies
We used KEGG, Reactome, BioCarta, WikiPathway, and Elsevier Pathway as our pathway analysis databases for this study. Then, using shared DEGs between COVID-19 and Smoking, we searched for highly expressed pathways. These pathways were then divided into functional groups for analysis as shown in Table 2. In all, 345 signaling pathways were initially associated with both disease and condition. The number of pathways was then reduced by using manual curation. Consideration is given to pathways with adjusted P-values under .05. Using P-value, we get unique numbers of every gene and easily identified them. Consequently, we obtained the 238 most important signaling pathways. Finally, we organized the pathways in ascending order based on the P-value.
Ontological analysis between COVID-19 and smoking.

Abbreviation: GO, gene ontology ; MHC, major histocompatibility complex; ER, estrogen receptor. TAP, transporter associated with antigen processing; NMDA, N-methyl-D-aspartate; COPII, cytoplasmic coat protein complex-II.
Figure 4 shows the top 20 pathways that were connected to COVID-19 and smoking status. The GO approach divides them into segments using the terms MF, BP, and CC, but we only consider the BP database of GO terminologies. Initially, 436 GO terms were discovered to be shared by smokers and COVID-19 patients. The terms with a P-value under 0.05 were then added as the most important GO terms. Between the conditions, 159207 GO terminologies are discovered as the most enriched GO terms. The top 20 most important GO terms of the BP between COVID-19 and Smoking are summarized in Figure 5. Biological (BP) and cellular processes are used in the GO approach.

Top 20 pathways are represented using a bubble plot that are associated with both conditions in transcriptomic analysis.

The figure illustrates the protein-protein interactions between COVID-19 and smoking.
Protein-protein interactions (PPIs) analysis
We utilized our enriched gene sequences for common diseases to generate putative PPI networks by using web-based visualization techniques. The Cyto-Hubba 50 was used to create the simplification PPI networks in order to determine the most significant hub proteins. Network Analyzer in Cytoscape was used to calculate the topological parameters, 51 as shown in Figure 5. The Maximal Clique Centrality (MCC), as well as BottleNeck algorithms, were used in conjunction with the CytoHubba plugin in Cytoscape to identify the highly linked hub proteins in the PPI network. Although more research is required to fully comprehend their roles, these recently discovered hub proteins may prove useful as therapeutic targets. This information confirms the inclusion of pertinent functional pathways and shows that the PPI subnetwork is present in our enriched gene sets. These hub proteins have been identified, but more research is needed to fully understand their functions. Figures 6A and B show the hub proteins that were discovered using the Bottleneck and MCC algorithms as previously mentioned. We obtained a total of 34 hub proteins; however, we have only reviewed 21 of them, only the top 3 being red, orange, and yellow. Using the MCC method, we were able to identify 17 hub proteins, of which 10 are considered to be the top hub proteins.

Hub proteins are identified using 2 different cyto-hubba algorithms called MCC and bottleneck to demonstrate the association between COVID-19 and smoking. (A) Bottleneck. (B) MCC.
In addition, using the BottleNeck algorithm, we found 17 hub proteins in total, with 11 of them being shown as the most important (marked as yellow, red, and orange colors). We have identified 14 important, distinct hub proteins across both algorithms, and they are as follows: C4A, SKIV2L, HLA-C, KRR1, UTP15, HLA—DQB, WDR46, UTP6, BCAR1, UTP3, NTSR1, NFKBIL 1, APOE, and NTS. Both algorithms share 7 of these proteins, including C4A, SKIV2L, HLA-C, KRR1, UTP15, and HLA-DQB. This information shows that the PPI subnetwork is present in our enriched gene sets and confirms the presence of relevant functional pathways. Although more research is required to fully understand their functions, the identified hub proteins may be helpful for treating STRING 51 targets via the network. Table 3 displays a summary of important hub proteins.
Summary of COVID-19 and smoking hub proteins identified by the algorithms of MCC and bottleneck.
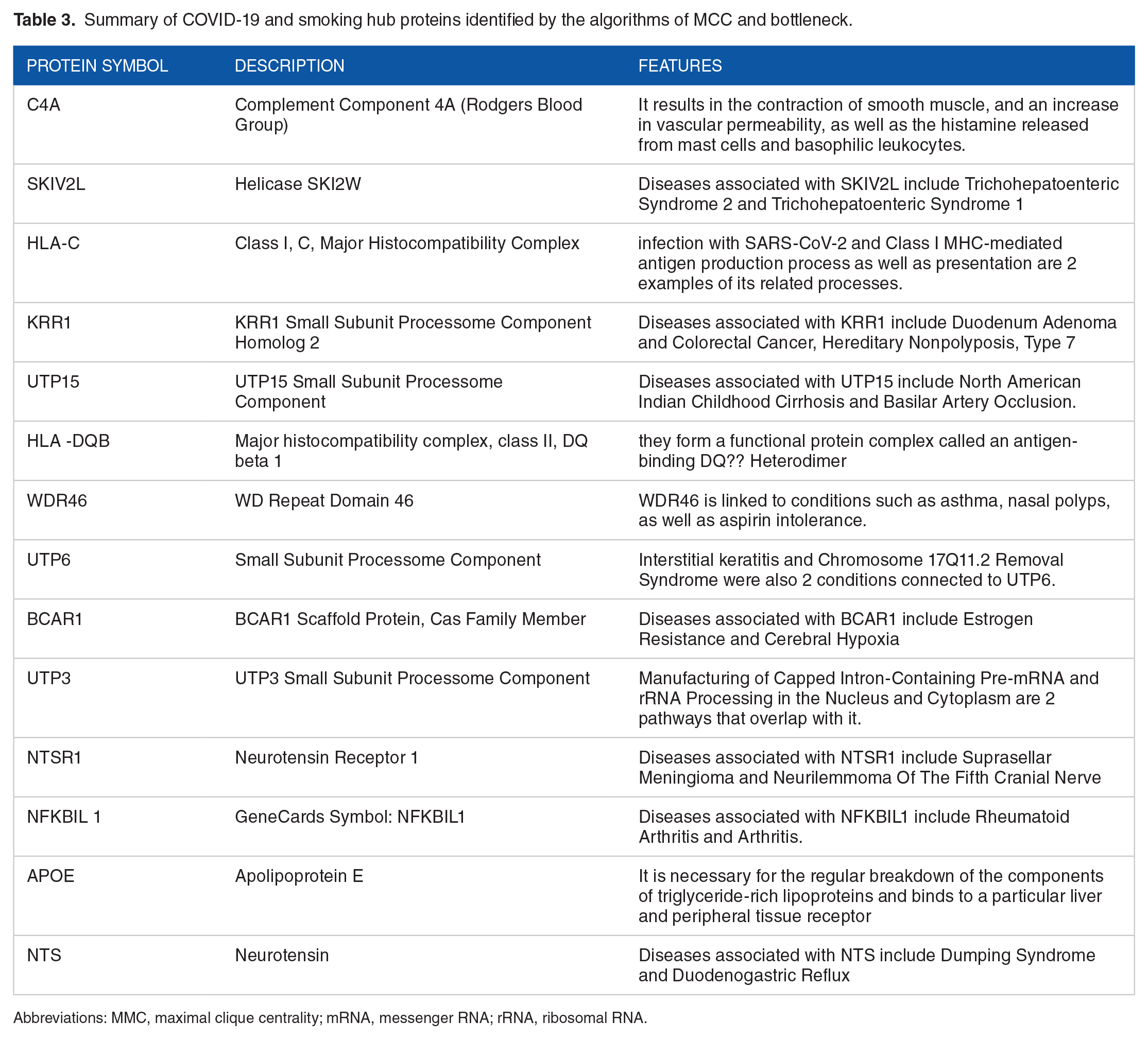
Abbreviations: MMC, maximal clique centrality; mRNA, messenger RNA; rRNA, ribosomal RNA.
Identification of transcriptional and post-transcriptional regulators of the differentially expressed genes
TFs are proteins that regulate genes as well as transcriptional expression over all living things. TFs play an important role to control gene and transcriptional expression in all living things. All cellular processes depend on the activity of transcription factors (TFs), 52 which is essential. We used Net-workAnalyst to analyze TFs and gene-microRNA interactions tools. 37 miRNAs are non-coding short RNA molecules that control gene expression after transcription. By using the ChIP-X 53 as well as JASPAR 54 databases, the TFs-Gene network was created. The network of gene-miRNA interactions was created using NetworkAnalyst and the TarBase 55 and miRTarBase 56 databases. Figure 7 visually represents the TF-Gene Interactions and the regulator genes are KLC3, SKIV2L, CHPF, CYP2W1, C4A, TFC19, EGFL7, BCAR1, NFKBIL1, PTGER1, PLXNB3, GRIN3B, APOE, GAS2L1, GP1BB, PITX1, ZBTB33, MAZ, NTSRI, TOX2, GP1BB, HLA-DQBI, IGFALS, NKFBIL1, BCAR1, CHPF, HINFP, USF2, GATA2, TAFA2A, E2F6, TFA2C, YY1, STAT1, FOXC1, STAT3, NFKB1, and RELA were identified as the key regulators of the identified DEGs. Gene interactions involving miRNA are shown graphically in Figure 8: C4A, SKIV2L, NFKBIL 1, CHPF, GRIN3B, IGFALS, GP1BB, GAS2L1, APOE, BCAR1, TRIM27, PLXNB3, EGFL7, NR2F1, ZBTB33, SP1, GLIS2, MYNN, MAZ, BCL11B TOX2, NFKB, TCF19, CHPF, CYP2W1, NPW, BCAR1, LMNTD2, IGFALS, NFIC, USF2, TFAP2A, E2F6, YY1, FOXCI, NFKBI, SREBF1, RELA, and GATA2.

TF-gene interactions showed using 2 different algorithms called ChEA and Jaspar to illustrate the linking between COVID-19 and smoking. (A) Encode. (B) JASPAR.

Gene miRNA is identified between COVID-19 and smoking using 2 different algorithms called TarBase and MirTarBase. (A) Tar-Base. (B) Mir-Tar-Base.
Drug-protein interaction
The protein–drug interactions for Smokers as well as a COVID-19 side effect and Common Genes are shown graphically in Figure 9 as follows: GRIN3B. The genes are connected to drugs called phenobarbital halothane, orphenadrine, secobarbital, atomoxetine, pen-tobarbital, acamprosate, gavestinel, tenocyclidine, dehydropiandroseterone, ketabemidone, glycine, L-glutamic acid, minacipran, and gabapentin.
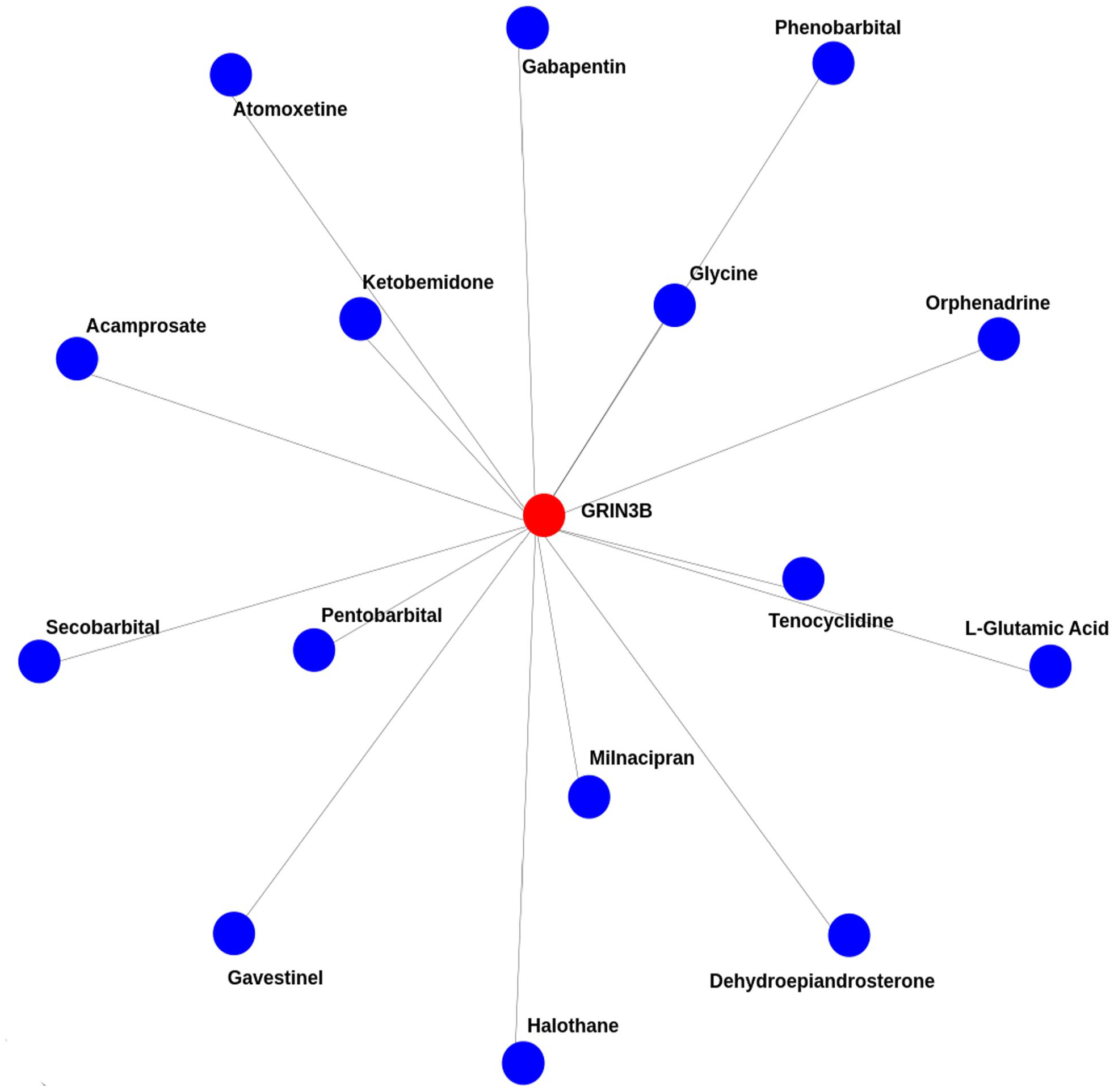
The figure shows the protein drug interaction between COVID-19 and smoking using protein drug interaction algorithm.
Discussion
COVID-19 provides a significant threat to the general people while smoking remarkably raises the risk of both COVID-19 and mortality. 57 Mainly, smoking is primarily responsible for suffering from acute COVID-19 consequences. 7 COVID-19 sufferers who smoke regularly are in danger of developing various complications or diseases. So, in our study, we use several bioinformatics methods to discover genetic links between COVID-19 and smoking. Furthermore, our predicted medications can be considered to treat COVID-19 patients with a smoking history and doctors will encourage their patients to abstain from smoking. Undoubtedly, this will reduce the risk that people would develop COVID-19 or other complications brought on by smoking in COVID-19 patients. The bioinformatics method helps us to understand the mechanisms of underlying diseases.
COVID-19 patients have C4A (Complement Component 4A) accumulation in the lung tissue58,59 and due to active smoking brain, C4A explication can be raised. 60 C4A, which are wide immunologic activators, are known to encourage a number of immune processes, such as immune cell chemotaxis as well as NETosis, reactive oxygen species, also the production of cytokines, inflammasomes, and eicosanoids. These immune processes are playing an important role in a number of respiratory damages in COVID-19 patients. 61 SKIV2L genes were observed to have significant associations with the presence of RPD, and the RPD rate was higher among patients with a history of smoking. 62 It illustrated that SKIV2L included in the RNA exosome-activating SKI complex, limits baseline type I IFN accounts, which all are instigated by RNA sensors in circumstances where SKIV2L is deficient. Moreover, SARS-CoV-2 replication is restricted by pharmacological prohibition of the SKI intricate. 63 It has been proposed that the enzymatic SKI complex subunit, SKIV2L, controls the IFN reactions by modulating RIG-I, which may indicate COVID-19 patient mortality. 64 Patients succumb to COVID-19 quickly because there aren’t any early IFN responses against SARS-CoV-2. 65
At first, we focused on 5 different pathways namely, the endothelial cell adhesion pathway, ER-phagosome pathway, neuroactive ligand-receptor interaction pathway, intestinal immune network pathway, and human cytomegalovirus infection pathway. In the endothelial cell, raised manifestation adhesion molecules are connected to COVID-19 patients. 66 Two types of infection that causes severe acute respiratory syndrome can trigger the release of cytokines, activating the endothelial cells pathway. This could result in vascular changes, which are endothelial dysfunction, pyroptosis, and thrombosis usually mentioned to COVID-19. 67 And in the vascular cell, patients with coronary heart disease who smoke experience higher plasma levels of adhesion molecule-1. 68 Furthermore, phagosome pathways, besides inflammasomes, were discovered in the lung infected by SARS-CoV-2.69,70 ER-phagosome pathway protein-binding AU-rich elements control mRNA stability in COVID-19 patients 71 as well as the phagosomal pathway was enriched in smokers. 72 Moreover, the KEGG-neuroactive ligand-receptor interconnection pathway demonstrated momentous relations with lung cancer related to smoking. 73 In addition, in COVID-19 patients, the neuroactive ligand-receptor interaction pathway is one of the important pathways of 5 remarkably high-regulated pathways. 74 According to molecular docking, 2 protein of coronavirus had a good affinity for the neuroactive ligand-receptor interconnection pathway. 75 Notably, the symptoms of COVID-19, which can be controlled by the calcium signaling pathway and neuroactive ligand-receptor interaction pathway, include myalgias, headaches, and abdominal pain. 76 COVID-19 sufferers could grow severe signs, namely high pathway, “intestinal-immune-network” for IgA production, and “T-cell-receptor signaling pathway.” 77 The immune systems of COVID-19 patients’ oral cavity and intestinal tract are controlled by serum IgA and secretory (s-)IgA. 78 The effect of smoking produces the small intestine’s mucosal IgA antibody. It was found that genes highly connected with the immune-network involved in IgA-production signaling pathways included in the end part of the tiny intestine after exposure to cigarette smoke. 79 And KEGG-pathway-enrichment easily concluded in 156 pathways associated with COVID-19, and the human cytomegalovirus infection pathway is one of them. 80 Human cytomegalovirus infection pathways are related to the regulation of inflammation of COVID-19 patients. 81 Also, smokers have a higher tendency to be infected by human cytomegalovirus infection pathway. 82
In the hub-protein interaction, we have validated total of 7 proteins with previous literature named WDR46, C4A, APOE, HLA-C, HLA-DQB1, BCAR1, and NTS. Smoking and WDR46 interaction may be responsible for impacting young people’s lung development, which may affect the development of lung cancer in adults. 83 Moreover, lungs from severe Significant C4a deposits were found in COVID-19 patients, which may indicate that complement plays a role in lung damage. 84 In COVID-19 patients with severe there is strong immunohistochemical staining for C4a as well as C3, and also MAC that colocalizes with SARS-CoV-2 nucleocapsid protein. 85 As well as, smoking expands the declaration of the C4A gene. 60 We also discover that smoking expands the declaration of the C4A in the brain. It’s interesting to note that smoking is linked to diffuse as well as epidemiological evidence, and also dose-dependent cortical thinning suggests that smoking increases the risk of developing schizophrenia. 60
According to previous the sensitivity and also cruelty of COVID-19 are associated with a number of genetic variables, including APOE. 86 In the case category, APOE-associated comorbidities were danger factors for intense COVID-19. Another in vitro research discovered that SARS-CoV-2 endocytic entry was increased in cholesterol-loaded cells using the cholesterol transport protein APOE. 86 The Apoe-isoform-dependent effect may aid in describing the elevated risk for severe COVID-19. By applying health data and also genetic data from the Biobank of the United Kingdom, current research has demonstrated where the Apoe E4/4 genotype grows the danger for intense COVID-19, in comparison to the ApoE3/3 genotype, independent of preexisting comorbidities, including cardiovascular diseases, type 2 diabetes as well as dementia, this suggests an association between ApoE4 and COVID-19. 87 Also, the genotype of APOE e4 may potentially be related to smoking cessation. 88 APOE genes are used in developing the danger of ischemic heart disease highly related to the active smokers. 89 In addition, HLA-C loci are crucial in controlling how severe COVID-19 disease is clinically. 90 HLA-C carriers are more likely to experience COVID-19’s severe clinical course. After Comparing with other HLA-alleles, it was highlighted that HLA-C has lower predicted binding sites for pertinent SARS-CoV-2 peptides. 91 HLA-C was remarkably linked to COVID-19 patients’ most severe disease, necessitating admission to an ICU. 92 And passive smoking and HLA DQB1 positive were risk factors for narcolepsy. 93 We postulated that let-7c-ADRB1-HLA-DQB1-AS1 interconnections are important in the development of smoking-related chronic obstructive pulmonary disease (COPD). 94 In people with the HLA DQB1 gene, passive smoking may raise the risk of progressing narcolepsy. 95 In COVID-19 patients, NTS may contribute to the severe aggravation of pulmonary edema, and microvascular clotting. 96 There are different kinds of chemokines and cytokines are attracted NTs which also raised in infections of SARS-CoV-2, recommending a potential role for NTs in the defense opposed to SARS-CoV-2. In contrast, SARS-CoV-2 can circumlocutorily activate the development of neutrophil extracellular traps (NETs) in COVID-19 by inducing CS and also down-regulating ACE2, which prevents NTs-infiltrations. 97 BCAR1 is associated with cigarette smoking and lung cancer. 98 In cases of adenocarcinoma or squamous carcinoma in former or current smokers, BCAR1-mRNA levels predicted a worse prognosis. Breast cancer with high BCAR1 levels has a poor prognosis and is more likely to relapse. 99 In addition, lung cancer patients with upregulated BCAR1 had a worse prognosis and had a big risk of distant metastasis, lymph node metastasis, and chemotherapy resistance. 100
For drug-protein interaction, by previous literature, we were able to validate a total of 7 drugs, namely halothane, atomoxetine, acamprosate, glycin, minalcipran, gabapentin, and dehydroepiandrosterone (DHEA). halothane is a volatile anesthetic agent. The antiviral effect of volatile agents is one of the most significant effects when considering the possibility that they can treat COVID-19 acute respiratory distress syndrome (ARDS). Exposure to halothane at a concentration of 2.2% mildly inhibited the replication of many animal viruses. 101 Where halothane inhibits the dimerization of the SARS-COV-2 encoded nucleocapsid protein. 102 Patients with attention deficit hyperactivity disorder (ADHD) who take non-stimulant medications (such as viloxazine, as well as atomoxetine, also clonidine, and guanfacine), have a cancer diagnosis or were in palliative care at all point before their index the COVID-19 virus. 103 Atomoxetine is, according to previous literature, remarkable growth in pulse as well as in blood pressure (BP). 104 In addition, COVID-19 patients typically take atomoxetine 10 mg once a day to treat their depression as well as attention deficit disorder, and the dose was gradually increased to 40 mg. 105 According to research, sodium acamprosate had a strongly prohibited task as opposed to 3CLpro from SARS-CoV-1, exposing an inhibition amount of more than 85%. 106 As well as, acamprosate, a modulator of human GABRA1 protein, is in Phase IV as a part of the combination drug acamprosate and escitalopram as components of treatment for alcohol abuse. 107 The US Food and Drug Administration (FDA) granted acamprosate permission in the United States in 2004 for the therapy of alcoholism. Both metabotropic glutamate receptors and N-methyl-d-aspartic acid channels are affected by acamprosate. 108 The use of acamprosate calcium can be resisted SARS coronavirus. 106 In another research, glycine is a non-essential amino acid that is being studied as a positive mitigator of cell damage and proinflammatory storms in COVID-19 patients. Two possible medications, glycine as well as pyridoxal, target the pathogenesis-related biomarkers of COVID-19 that we identified. 109 The elevated thrombotic risk linked to COVID-19 may be practically reduced by high-dose glycine. 110 Potential interactions between COVID-19 treatments, milnacipran and vortioxetine, will be deemed less risky. 111 Using the L1000FWD web-based application, analysis of medications possibly helpful for treating COVID-19 patients who also have T2D was done on the 35 high-regulated and 14 low-regulated genes shared by COVID-19 infected pancreas organoids and Type-2 Diabetes islets. Vemurafenib, milnacipran, and abstain-analog were the 3 leading medications with the highest anti-similar signal. 112 In this select group of patients with chronic persistent pain for a year or longer after knee arthroplasty, milnacipran reported decreased pain and some indications of functional development, demonstrating the necessity for well-powered investigations. 113 In 2013, discernible difference in efficacy and tolerability. 114 Milnacipran has been demonstrated to improve transmission in the descending pain pathways, reducing pain intensity and stiffness and enhancing function. 115 We utilized 50 mg of tramadol, 100 mg of gabapentin every night, 30 mg of duloxetine twice a day, and ketorolac as needed to treat pain in a clinical trial. The patients who are suffering from COVID-19” pain crisis” had resolved by week 4, but still had acute pain scored as 4-6/10. The patient experienced ongoing myalgia as well as numbness, tingling, and weakness. We retained the dose of duloxetine at 30 mg 2 times daily while raising gabapentin to 200 mg every night. By week 6, the patient’s neuropathic discomfort had disappeared. 116 The gabapentin dosage was raised to 400 mg per night. Moreover, a second negative result came from the SARS-Cov-2 test. Though gabapentin is used in COVID-19 patients, gabapentin increases the risk of kidney injury in COVID-19 patients. So, adjust the gabapentin dose based on renal function. 116 As information from the previous study, DHEA sulfate has a function in COVID-19 prognosis, and therapy 117 and DHEA is used to treat ARDS in COVID-19 sufferers. 118 signs of COVID-19 that are severe seem to be more likely in people with higher levels of circulating androgens. To see if low serum testosterone levels have an impact on COVID-19 infections and their severity. Dehydroepiandrosterone is being used as a booster of COVID-19 treatment. 119 Considering the COVID-19 epidemic, DHEA is a potent inhibitor of glucose-6-phosphate dehydrogenase (G6PD), because it has been demonstrated that a decrease in G6PD activity makes human cells more susceptible to coronavirus 229E infections. 117 And, DHEA, a recognized antiviral drug, levels play an important role in newborns’ defense against COV-19. 120 A sigma-1 receptor agonist, DHEA, is a testosterone precursor. Dehydroepiandrosterone, the substances for the sigma-1 and sigma-2 receptors, were effective inhibitors of SARS-CoV-2 replication. 121 Furthermore, DHEA may counteract the anti-inflammatory effects of glucocorticoids used to treat severe COVID-19 complications. 122 Dehydroepiandrosterone immune response mechanism, which could provide new therapeutic insights for the subset of COVID-19 patients who experience cutaneous manifestations. 123 All of our results have been validated against the scholarly literature as much as feasible. For this reason, there is hush opportunity for more in vivo and in vitro level research as it was not possible. A physician may advise current smokers to quit based on these results to reduce the severity danger of COVID-19. It’s possible that the pharmaceutical industry, using the resulting chemical molecule, may create medications to treat people with COVID-19 with a smoking history. It is important to emphasize that the smaller sample sizes of the datasets are a drawback of our research. This analysis didn’t take into consideration age, sex, race, or any other possibly relevant factors. Therefore, additional validation is required to carefully examine the biological remarkable of the problem as indicated in this work as shown in Table 4.
Previous research has validated the identified potential target genes or biomarkers that are related to smoking as well as COVID-19.

Conclusion
In this study, we used several bioinformatics approaches to learn more about the connection between smoking and the intensity of COVID-19. The transcriptomic analysis of our study revealed 37 shared molecular marker DEGs between T2D and smokers. Whereas 7 genes (WDR46, C4A, APOE, HLA-C, HLA-DQB1, BCAR1, and NTS) are validated using previous studies. Signaling pathways such as the Endothelial Cell adhesion pathway, ER-phagosome pathway, neuroactive ligand-receptor interaction pathway, intestinal immune network pathway, and human cytomegalovirus infection pathway is also verified using published literature. Also, we have validated 7 drugs named halothane, atomoxetine, acamprosate, glycin, minalcipran, gabapentin, and DHEA. There are several surprising new findings that open up possibilities for investigation. Finally, our research recommends that smokers should abort smoking to reduce the terrible impact of COVID-19. However, using our study based on drug prediction results, it would be possible to invent new drugs for COVID-19 patients with a smoking history.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Md Anisur Rahman, Md Al Amin, Most Nilufa Yeasmin, Md Zahidul Islam: data collection, conceptualization, methodology, software, validation, formal analysis, investigation, resources, data curation, writing—original draft preparation, writing—review and editing, visualization, supervision, project administration. All authors contributed to the article and approved the submitted version.