
Abstract
Prediction of key features of protein structures, such as secondary structure, solvent accessibility and number of contacts between residues, provides useful structural constraints for comparative modeling, fold recognition, ab-initio fold prediction and detection of remote relationships. In this study, we aim at characterizing the number of non-trivial close neighbors, or long-range contacts of a residue, as a function of its “topohydrophobic” index deduced from multiple sequence alignments and of the secondary structure in which it is embedded. The “topohydrophobic” index is calculated using a two-class distribution of amino acids, based on their mean atom depths. From a large set of structural alignments processed from the FSSP database, we selected 1485 structural sub-families including at least 8 members, with accurate alignments and limited redundancy. We show that residues within helices, even when deeply buried, have few non-trivial neighbors (0–2), whereas β-strand residues clearly exhibit a multimodal behavior, dominated by the local geometry of the tetrahedron (3 non-trivial close neighbors associated with one tetrahedron; 6 with two tetrahedra). This observed behavior allows the distinction, from sequence profiles, between edge and central β-strands within β-sheets. Useful topological constraints on the immediate neighborhood of an amino acid, but also on its correlated solvent accessibility, can thus be derived using this approach, from the simple knowledge of multiple sequence alignments.
Keywords
Introduction
Among the set of relatively simple principles that governs the three-dimensional structures of globular protein domains (Chothia, 1984), two are of obvious importance: i) the masking of a large part of the main chain polarity through the establishment of hydrogen bonds between the amide protons and carbonyl oxygens (mainly within α-helices and β-sheets) and, ii) the hydrophobic effect, underlying the formation of hydrophobic cores of globular domains. In this context, we have highlighted several years ago that strong hydrophobicity has to be conserved in some key positions of a given fold, which were called “topohydrophobic” positions (Poupon and Mornon, 1998; Poupon and Mornon, 1999; Poupon and Mornon, 1999; Poupon and Mornon, 2001). Within a typical globular domain, a third of amino acids belongs to a clear hydrophobic group (VILFMYW), but only a half of these strong hydrophobic amino acids occupies “topohydrophobic” positions (Poupon and Mornon, 1998; Poupon and Mornon, 1999; Poupon and Mornon, 1999; Poupon and Mornon, 2001), which are mainly located within α- and β- regular secondary structures.
“Topohydrophobic” positions have noticeable features, as observed from a comprehensive analysis of structural alignments and their associated three-dimensional structures: i) the amino acids in these positions are much more buried than those occupying “non-topohydrophobic” positions (Poupon and Mornon, 1998); ii) the side chains of these amino acids are markedly less dispersed from one domain to another (though belonging to the same fold), than those located at “non-topohydrophobic” positions (Poupon and Mornon, 1998; Poupon and Mornon, 1999); iii) they constitute a continuous network of positions in close contact, matching well the inner part of the hydrophobic core (Poupon and Mornon, 1998; Poupon and Mornon, 1999); iv) they are mainly occupied by amino acids constituting the folding nuclei (Poupon and Mornon, 1999).
Identification of these “topohydrophobic” positions from the knowledge of sequence data only is possible in practice if an accurate alignment of a small number (e.g. 5 to 8) of sufficiently divergent sequences sharing the same fold (e.g. in the 15–25% sequence identity range) can be performed. From sequence data only, amino acids of crucial importance for the considered fold can be thus highlighted, thereby providing topological constraints at long distance along the sequences, which can be useful in a general way to understand topological features of the protein universe (Lindorff-Larsen et al. 2005).
In the present study, we refine and extend the concept of “topohydrophobic” positions, by introducing a generalized topohydrophobic index, which evaluates at each position of a given sequence alignment the fraction of amino acids belonging to the hydrophobic group. We then wish to characterize the number of non-trivial close neighbors of each position of a multiple alignment, depending on this generalized topohydrophobic index deduced from current evolutionary profiles and on the associated predicted secondary structure state. The non-trivial close neighborhood of a residue, which can also be defined as non-local or long range contacts, is the set of amino acids sufficiently distant in the 1D sequence but close in the tertiary structure of the considered protein domain. Residues known to be in local proximity (e.g. covalence and α or β local chain neighbors) are excluded from this set.
In order to define the foundations for predictive studies, we first perform a comprehensive analysis on the basis of accurate reference alignments, selected from structural databases. Hence, we consider a large set of structural alignments allowing good statistics and only focusing on regular secondary structures that are at the building blocks of protein globular domains. Thus, the core blocks defined in this way only include regions aligned with maximal reliability. The topohydrophobic index is based on the natural partition of amino acids in two groups, considering the mean atom depth associated with each kind of amino acid (Pintar et al. 2003; Pintar et al. 2003). This value is indeed closely related to the mean hydrophobicity, and provides a clear separation between hydrophobic residues and the other ones.
The present analysis significantly differs from previous estimations of absolute contact numbers of residues from amino acid sequence data (Fariselli and Casadio, 2000; Ishida et al. 2006; Kinjo et al. 2005; Pollastri et al. 2001; Pollastri et al. 2002; Yuan, 2005). Indeed, these studies generally consider all contacts in a large sphere (typical distance cut-off of 12 Å between Cβ atoms), whereas we focus here on the mean local non-trivial neighborhood of a position within both kinds of regular secondary structures (α-helices and β-strands) using multiple alignments and a short distance cutoff of 7 Å between Cα atoms. Consequently, the number of predicted neighbors is considerably smaller, in the range of 0 to 6, instead of typically 0–50, as described in previous works. Our study also differ from those devoted to the prediction of long range contact maps (e.g. Punta and Rost, 2005), as these do not generally focus on the quantification of these contacts with respect to the secondary structure and to the evolutionary hydrophobicity profile of the considered residue.
We show here that an informative neighborhood of residues can be highlighted from sequence data, which differs between helices (often 0 to 2 such neighbors) and strands (mainly 3 to 6 neighbors). Moreover, a clear multimodal behavior of strands can be observed, with a first main state around three neighbors (tetrahedral arrangement), and the other one around six neighbors (two tetrahedra sharing a vertex). This multimodal behavior allows the distinction between central and edge β-strands. Given the high accuracy reached by secondary structure predictors using multiple alignments (e.g. Frishman and Argos, 1997; Jones, 1999; Pollastri and McLysaght, 2005; Rost and Sander, 1995; Thompson and Goldstein, 1997), the present study offers the possibility of acquiring a good quality information to predict tertiary structures from sequence data only, using a minimal number of parameters.
Methods
Datasets and reduction of redundancy
The structural alignments used in this study provide enough data to obtain accurate results, while still supporting a structural relevance. Structural alignments performed and/or extensively corrected by human expertise, as those used for the previous description of “topohydrophobic” positions (Poupon and Mornon, 1998), furnish particular good data; however, due to the considerable increase of structural data, such an expert-based procedure is now unconceivable for analysis on a large scale.
Among the main available databases of structural alignments (e.g. BaliBASE (Thompson et al. 1999; Thompson et al. 1999), HOMSTRAD (Mizuguchi et al. 1998), PALI (Balaji et al. 2001), FSSP (Holm and Sander, 1994)), only FSSP (after
Combining both thresholds (SI = 50% and Z ≥ 4), we obtain a database of 1721 sub-families of at least 8 members, including a total of 98 436 sequences, 2876 sequences being distinct from each other. Figure 2A summarizes this process (steps 1 to 3). Step 4 considers a composition identity (CI) threshold between families (0.5, 0.5) (see below and Fig. 2B).

The original FSSP alignments are reformatted according to the following information: sub-family name and PDB accession number of the leader sequence, number of members (≥8), PDB accession numbers of these members, associated structural FSSP Z indexes, alignment length, corresponding aligned sequences and aligned secondary structures (assigned through DSSP (Kabsch and Sander, 1983)). In addition, 3D coordinates of α-Carbons and solvent accessibilities calculated by DSSP (Kabsch and Sander, 1983) are reported for each residue. Figure 3 shows a typical file for a family of eight members.

Amino acid classes
The large dataset of reliable multiple alignments constituted here remains however considerably too small to consider the twenty different amino acids in each work position. The clustering of amino acids into a limited number of classes is thus necessary. Usually, three to six classes may be rationally defined (e.g. VILFMYW for the strong hydrophobic class, mainly present within the internal sides of regular secondary structures, GPDSN as main loop-forming residues and ARC-QTEKH for the intermediate class (Callebaut et al. 1997; Hennetin et al. 2003)). Here, we consider a simple partition into two classes, derived from a continuous scaling of the 20 amino acids with respect to their mean atom depth, as defined from a representative set of globular proteins (Pintar et al. 2003; Pintar et al. 2003). Mean atom depth indeed allows the sorting of the 20 amino acids in two distinct groups: IVFLWMCYA (G1) and HTGSPNRQDEK (G2) (Fig. 4). This classification shows good agreement with mean amino acid burying values, defined through Voronoï tessellations on representative sets of globular domains (Soyer et al. 2000). The two main groups G1 (mainly hydrophobic amino acids) and G2 (mainly neutral and hydrophilic amino acids) gather 44 and 56% of the total number of amino acids, respectively. The amino acids of group G1 are similar to those that were considered hydrophobic by other studies dedicated to long-range contacts (e.g. Punta and Rost, 2005).

Work Positions
We name “work positions” positions in the multiple alignment for which at least 8 amino acids are aligned. The consideration of this absolute number, rather than a relative proportion of all aligned sequences, allows the handling of representative subsets of these alignments, while ignoring positions in which gaps are predominant.
Generalized topohydrophobic index
Each work position is characterized by its percentage in amino acids belonging to the G1 group. We name it generalized topohydrophobic index or y1, because it records the proportion of hydrophobic amino acids (G1) occupying the position. Distributions of the y1 parameter are plotted within histograms, according to grouping intervals of 1/8 as a reference to the minimal number of amino acids (8), which have to be present in a work position to be considered.
Major secondary structure
We choose to take into account only work positions in which a same secondary structure is sufficiently conserved (at more than x%). Figure 5A shows the number of work positions as a function of this threshold x. We consider that x ≥ 75% offers an acceptable compromise, ensuring that work positions are structurally relevant according to the secondary structure conservation and keeping enough data to perform a large-scale study. Figure 5B shows the distribution of work positions in the different secondary structures as a function of the generalized topohydrophobic index y1.

Mean solvent accessibility of a work position
Relative accessibilities are computed starting from the absolute accessibilities provided by DSSP (Kabsch and Sander, 1983). The standard accessible surfaces in Å 2 for residues are derived from canonical G-X-G configuration calculations by Shrake and Rupley (Shrake and Rupley, 1973): A: 124 / C: 94 / D: 154 E: 187 / F: 221 / G: 89 / H : 201 / I: 194 / K: 214 / L: 198 / M: 215 / N: 161 / P: 150 / Q: 190 / R: 244 / S: 126 / T: 152 / V: 169 / W: 265 and Y: 236. Relative accessibility of a work position is the mean value of the relative accessibilities of its residues.
Non-trivial neighbors
The non-trivial neighborhood of an amino acid can be described from the known atomic coordinates.
Two amino acids are defined as non-trivial neighbors if their Cα are separated by less than 7 Å (Tudos et al. 1994) and if they are distant in sequence from more than 6 residues (Fig. 6). The mean number of neighbors for a work position is defined as the average number of non-trivial neighbors of the amino acids belonging to that position. An even better way to consider the amino acid neighborhood, which is independent of a cutoff threshold value, would have been to use a description through pondered Voronoï tessellations (Angelov et al. 2002; Dupuis et al. 2005; Dupuis et al. 2004; Soyer et al. 2000). However, this description is prohibitively time-consuming and thus out of scope for a large-scale study.
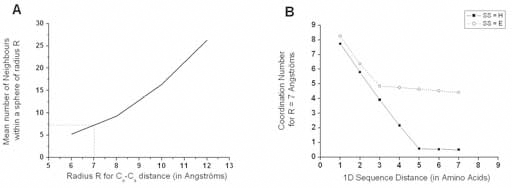
Neighboring definition. A. Mean number of neighbors within a sphere of radius R, as a function of Cα-Cα distance R, calculated on the FSSP-derived bank. For R = 7 Å (a value generally retained to characterize close neighborhood of an amino acid), the mean coordination number is between 7 and 8. B. Evolution of the mean coordination number for R = 7 Å as a function of the sequence distance D, expressed in amino acids. For D = 1, all contacts are taken into account and the mean values are close from each other for strands (E) or helices (H). Above D = 2, behaviors of strands and helices differ, as strands assemble to form sheets with a high and constant mean number of neighbors (~4.5), while helices only show a small mean value of ~0.5 when D ≥ 4. For the E and H states, we consider that beyond D = 6, neighbors are only non-trivial ones.
Results
Dataset
A set of benchmark alignments is selected as described in the Methods section, in order to estimate the number of long-range (or non trivial) contacts of amino acids, with respect to the general topohydrophobic index deduced from the multiple sequence alignment and to the associated secondary structure. The dataset considered here includes 1485 sub-families (31 327 sequences and 2727 distinct amino acids chains) with at least 8 members (mean number 20) and sharing no more than (0.5, 0.5) composition identity, a parameter that was introduced in order to avoid redundancy between subfamilies. In a given family, pairwise sequence identity is necessarily less than 50% and quite always far below (mean 8.3 %) and the members have a confident structural alignment quality (Z) of at least 4 (mean 7.3) with respect of the leader sequence of the family. It is worth noting that all proteins sharing a same fold, fulfilling the selected sequence identity and structural alignment quality criteria described above, are not clustered into a unique family. Some sub-families described above are subsets of proteins possessing at least one domain with a given fold. This distribution in several sub-groups is directly dependent on the initial FSSP dataset and to the selection procedure. For example, some members of the family shown in Figure 3 (family 1mai—Pleckstrin Homology (PH) fold) are found in eight other families with a PH fold domain. However, the alignments well cover the known universe of globular domains, and are thus representative of the structural conservation and diversity within proteins.
We analyze the main features of “work positions” in multiple alignments (see definition in the Methods section), for which more than 75% of the residues share the same secondary structure. As structural superimpositions and secondary structure assignments were automatically performed, local mismatches may occur. However, these mismatches only constitute a marginal fraction within the final alignments obtained after filtering of the initial dataset. Only 8% of the 97 000 retained work positions exhibit more than one H/E discrepancy and thus only constitute a background noise, which do not sensibly modify the main results of this study. The good quality of solvent accessibility predictions, which are directly performed on our filtered database of structural alignments (see below) and are similar to results obtained with other methods (Gianese et al. 2003; Pascarella et al. 1998; Rost and Sander, 1994; Thompson and Goldstein, 1996), further supports the overall structural relevance of work positions.
The partition of amino acids in two groups G1 (IVFLWMCA) and G2 (HTGSPNRQDEK), as introduced in the Methods section, and the distribution of group compositions in 1/8 lead to 9 distinct topohydrophobic y1 values (0, 0.125, …, 1), which can describe a work position. 27 classes of work positions (X, y1) can thus exist, combining y1 and X, the major secondary structure (X = helix, strand or coil). The 27 classes are often largely represented in the bank. The less populated classes are the limit cases, consisting in fully hydrophilic strands (Strand, 0) and fully hydrophobic coils (Coil, 1) (462 and 296 work positions, respectively; Fig. 5B). We principally consider the 18 classes of work positions associated with regular secondary structures (X = H or E; 60 021 and 36 830 work positions, respectively).
Positions within helices
Relative solvent accessibility
Figure 7A illustrates the behavior of the mean relative solvent accessibility in helix work positions within multiple alignments, as a function of the generalized topohydrophobic index y1, ranging from 0 to 1. As expected, the mean relative accessibility to solvent diminishes when y1 increases. We also consider the individual behaviors of G1- and G2-residues. We observe that the G1- and G2-values depend on the y1 value of the work positions, and both diminish when y1 increases. The two curves are quite parallel for the two groups, with the G1 mean values smaller, as expected, than the G2 ones. The distribution of mean relative accessibilities around the mean values, shown in Figure 7A, is illustrated in Figure 8A. For very low y1 values (low hydrophobicity), the mean relative accessibilities are distributed according to a Gaussian-like rule centered on 0.45 and, as y1 increases, this curve smashes towards the origin, with a mean below 0.1 for 95% of the 1977 totally hydrophobic work positions (y1 = 1). For y1 = 0 (fully neutral or hydrophilic positions), a small peak, indicated by a star, reveals the existence of buried positions. It likely corresponds to salt bridges, and more generally to pairs of side-chains in mutual neutralizing polar contacts within globular cores. This observation moreover provides indirect biophysical support to the data quality of the FSSP-derived bank.

Helices. A. Mean solvent accessibility for helices, as a function of the composition of work positions. When positions have a high topohydrophobic index y1, the G2 class adopts a similar behavior as the G1 one, constrained by fold requirements.

Helices. A. Distributions of work positions according to mean relative solvent accessibility and hydrophobicity for α regular secondary structures. Star indicates an exceeding value, likely resulting from salt bridges and mutually neutralizing pairs of hydrophilic amino acids within protein cores. B. Distributions of work positions according to the mean number of non-trivial neighbors and hydrophobicity in α regular secondary structures. C. Typical single inter-helix contact found in 1mai, between H120 and A21.
Number of non-trivial close neighbors
The number of non-trivial close neighbors (Fig. 8B) shows a symmetrical behavior compared to the relative accessibility (Fig. 8A). The number of non-trivial neighbors of work positions within helices increases as hydrophobicity rises from y1 = 0 to y1 = 1, but is rarely greater than 2, even for completely buried positions (mean accessibility < 0.1), within the internal sides of helices. This mainly results from the principal occupancy, in such configurations, of the close neighborhood by trivial neighbors, which restrains the free space for external residues, and from the convex geometry of α-helices, roughly cylindrical, with a large dispersion of side chains. G1 and G2 groups are both concerned by this increase of the number of non-trivial neighbors (Fig. 7B). Work positions with high hydrophobicity within helices mainly establish contacts with other helices (Fig. 7C). Moreover, these contacts mainly involve G1 amino acids within the hydrophobic core (data not shown). Figure 8C illustrates such a situation.
Positions within Strands
A similar investigation was performed for work positions associated with β-strands (Figs. 9 and 10). The most striking result for β-strands is a strong increase of the number of the non-trivial first neighbors and a clearly multimodal distribution observed for almost all y1 values, and in particular for the less hydrophobic ones (low y1 values). The weakly populated mode, centered on approximately one neighbor, is likely associated with highly external positions at the extremity of some strands. The two other modes (near 3 and 6 neighbors) are likely to correspond to external (edge) and internal (central) positions of strands within β-sheets, respectively. Indeed, the second mode (around 3) mainly relies on the architecture of β-strands within sheets, where side chains in
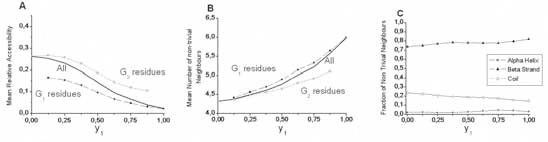
Strands. A. Evolution of the mean relative solvent accessibility for β-strands, as a function of hydrophobicity of work positions. B. Evolution of the mean number of non-trivial neighbors; as a function of the composition of β work positions. C. Partners of β-strand work positions. At high topohydrophobicity, strand-strand and particularly G1-G1 contacts dominate in β-sheets.

Strands. A. Distributions of work positions according to mean relative solvent accessibility and hydrophobicity for β regular secondary structures. The presence of salt bridges and hydrophilic pairs likely account for the value indicated by a star, as for helix positions.
positions i, i + 1, i + 2 in one strand occupy a roughly equilateral triangle. This triangle constitutes the basis of the interaction with another amino acid j of a neighboring strand, linked to the “i” strand through canonical main chain H-bonds. These four residues constitute a more or less deformed tetrahedron (distance between Cβ ~6.2 Å), which represents the basic unit of compact packing of similar sized spheres (Fig. 10C). The third mode (around 6) mainly corresponds to a geometry with two tetrahedra (one strand sandwiched by two others) sharing a vertex, which has 6 first non-trivial neighbors (Fig. 10C). Many deviations from this ideal scheme occur and tend to flatten the Gaussian distribution. As for helices, the number of non-trivial neighbors increases with hydrophobicity of a work position (Fig. 9B) and strand non-trivial neighbors are very often found within other strands (Fig. 9C). The present study quantifies this behavior and offers the opportunity to gain information on the probable participation of an amino acid in an internal or external strand position, through the only knowledge of multiple sequence alignments.
Influence of Fold Classes
The dataset is large enough to estimate the putative influence of fold classes on some parameters. Four main classes, as described in the SCOP classification (Murzin et al. 1995), were considered (all-α (297 sub-families), all-β (370 sub-families), α/β (530 sub-families) and α + β (131 sub-families)). One can expect that differences in the tertiary structures between the four fold classes are reflected in the level of hydrophobic contacts, involving residues of the G1 group, and in particular in positions with a high topohydrophobic index (y1 = 1). Hence, one can observe that the mean number of non-trivial neighbors belonging to the G1 group for strand work positions with a high topohydrophobic index is sensibly higher for the α/β class than for the three others (4.51 versus 4.02 (α), 3.25 (β) and 3.79 (α + β); Fig. 11). This is all the more noticeable than the total number of non-trivial neighbors of strands work position with a topohydrophobic index of 1 is rather constant (Table 1). A hypothesis to explain such a behavior is that a larger number of fully hydrophobic work positions with a structural role exist in the α/β and even α classes, but this remains to be further investigated. Furthermore, one can note that better performance of programs for the prediction of long-range contacts are reported by at least two studies for this same α/β class (MacCallum, 2004; Punta and Rost, 2005).
Contacts achieved by strand work positions with topohydrophobic index y1 = 1


Mean number of observed G1 non-trivial neighbors within the main fold classes.
Discussion
The prediction of non-trivial neighborhood, or long-range contacts, from protein sequences is of particular interest to improve comparative modeling and to enhance fold recognition and ab-initio fold prediction. It can also help to detect remote relationships between protein sequences and to solve experimental structures. Contact prediction methods have received much attention during the last decade and often combine the evolutionary information available from multiple alignments and the prediction of secondary structures. They can be roughly classified in two non-exclusive categories: statistical correlated mutations approaches (see for examples Halperin et al. 2006; Kundrotas and Alexov, 2006) and machine-learning approaches (see for example Punta and Rost, 2005). While most methods aimed at predicting contact maps, several other approaches have been developed to estimate the total number of contacts (Fariselli and Casadio, 2000; Ishida et al. 2006; Kinjo et al. 2005; Pollastri et al. 2001; Pollastri et al. 2002; Yuan, 2005), but these generally define large numbers of coordination, including trivial neighborhood, and rarely link these numbers to the topological and evolutionary features of the region which includes the concerned residue.
Our analysis outlines the relationship between the mean number of non-trivial neighbors and a topohydrophobic index, which relies on the mean hydrophobicity of a position within a multiple alignment of sequences, as a function of the secondary structure. The topological data we collected here might be used in a predictive perspective, as secondary structures can currently be predicted with a good accuracy using multiple alignments (see for example Rost and Sander, 1993). As noticed in earlier studies (Punta and Rost, 2005), the performance of the various estimations that can be made on the long-range contacts directly depends on the quality of the evolutionary profiles, which have to be large and to contain divergent sequences to furnish accurate information.
The original result of this study is that different behaviors relative to non-trivial neighbors can be observed for helix and for strand residues, and among strands, for central and edge β-strands. Starting from these observations, the prediction of the topological nature of β-strands can be approached using classification methods like decision trees (see Supplementary data 1). Briefly, using parameters such as the length of the strand, its mean hydrophobicity and periodicity of G1 and G2 residues, combined with topohydrophobic index, decision trees lead to an accuracy of 80% for the prediction of edge/central positions within β-sheets (Supplementary data 1). Although it is difficult to compare methods using different datasets for training and prediction, this approach appears to achieve a prediction accuracy similar to the one obtained by Siepen and coworkers (Siepen et al. 2003), which is based on the use of support vector machine (SVM) and decision trees.
The use of the topohydrophobic index, combined with information on the nature of secondary structures, the group (G1 or G2) to which the residue belongs, as well as environmental parameters, describing the local periodicity, also allows the prediction of the relative solvent accessibilities of a residue within a work position into two or three states models (exposed, intermediate and buried; see Supplementary data 2). In the ideal case, when secondary structures are “known”, solvent accessibility predictions using this methodology led to Q2 of 79% (16% threshold) versus 75% for other methods tested on the same dataset and based on neural networks (Rost and Sander 1994) or probability profiles/ support vector machines (Gianese et al. 2003) and to Q3 of 65% versus 58% for the same other methods (9–36 % threshold). On the one hand, the accurate prediction of solvent accessibility using generalized “topohydrophobicity” provides additional constraints on informative positions of a sequence (the work positions). On the other hand, these results further support the intrinsic quality of the dataset used for this study.
The present analysis shed light on important geometrical and topological parameters that can help to understand protein sequence-fold relationships. It appears of particular interest that the dichotomy (hydrophobicity—hydrophilicity) between only two nearly equally populated classes of amino acids provides a very simple way to derive useful and often accurate topological data, that can be useful for protein fold recognition.
Footnotes
Acknowledgments
G.F. acknowledges a PhD grant of the “Direction Générale de L'Armement”.