
Abstract
In bioinformatics, multiple sequence alignment (MSA) is an NP-hard problem. Hence, nature-inspired techniques can better approximate the solution. In the current study, a novel biogeography-based optimization (NBBO) is proposed to solve an MSA problem. The biogeography-based optimization (BBO) is a new paradigm for optimization. But, there exists some deficiencies in solving complicated problems such as low population diversity and slow convergence rate. NBBO is an enhanced version of BBO, in which, a new migration operation is proposed to overcome the limitations of BBO. The new migration adopts more information from other habitats, maintains population diversity, and preserves exploitation ability. In the performance analysis, the proposed and existing techniques such as VDGA, MOMSA, and GAPAM are tested on publicly available benchmark datasets (ie, Bali base). It has been observed that the proposed method shows the superiority/competitiveness with the existing techniques.
Keywords
Introduction
More than three amino acid sequences or protein sequence alignment at a time is called multiple sequence alignment (MSA). MSA is the most important tool to solve biological problems. We can solve lots of problem in biology by using MSA. MSA helps to predict the secondary and tertiary structures of RNA and proteins.1,2 We can reconstruct phylogenetic trees using MSA, which can predict the function of an unknown amino acid by aligning its sequences with some other known functions. We can also find similarity of the sequences using MSA, which can help to define similarity in functions and structures.3,4 In order for an MSA to be valid, entire sequences in the multiple alignments must have a common origin. The goal of MSA is to maximize the matching of protein or amino acid as far as possible. 5 Therefore, MSA is an important problem in bioinformatics to study the genetic and phylogenetic relationship. There are several methods to solve an MSA problem in the past.
The MSA problem can be solved and an optimal alignment can be achieved by using dynamic programming (DP). DP uses a scoring function that contains a large domain. In 1970, Needleman and Wunsch 6 proposed the use of DP algorithm to solve the problem of two sequence alignments. But the problem behind the use of DP is that when the number and length of sequence are increased, its complexity also increases in an exponential manner. Then, the MSA problem becomes NP-hard. Since complexity is the main constraint for the computer to solve any problem, we have to maximize the matching of protein or amino acid sequence in limited time or less complexity. This is the major reason why researchers switch to other methods.
The MSA problem can be also solved using progressive method. The progressive approach takes less complexity in terms of time and space for solving an MSA problem.7,8 According to progressive alignment method, initially align more similar sequences and then incrementally align more divergent sequences or group of sequences in the initial alignment. The standard representative of progressive methods is CLUSTALW. 9 In the first step, according to this approach, we have to assign the weight of each pair of sequences in a partial alignment. We assign small weight for most similar sequences and big weight for most divergent sequences. After that, we take substitution matrix that defines the score between two residues of protein sequence based on similarity. Two types of gap have been introduced in the third step. The first one is residue-specific gap and the second one is locally residue gap penalties. In the fourth step, gap that has been introduced in early position receives locally reduced gap penalties to encourage the opening gap at these positions. These four steps are integrated into CLUSTALW, which is freely available. Progressive alignment method performs better for MSA package in terms of accuracy and time. Even this method has some limitation. The problem behind this method is dependency on initial alignment and choice of scoring scheme. In other words, we bound that to align more similar sequences in the initial stage. If we have not aligned more similar sequences in the initial stage, then the solution may be trapped in local optima. An iterative method is another option for solving MSA.
An iterative method does not depend on initial alignment because it starts with initial alignment and improves the solutions per iteration until no more improvement is possible. The main objective of the iterative approach for MSA is to globally improve the quality of a sequence alignment. There are some iterative and stochastic approaches for MSA (for example, simulated annealing10,11). Hidden Markov Models Training (HMMT) 12 is based on a simulated annealing process. The problem behind the solution recommended by these methods may be trapped in local optima.
Evolutionary algorithms13,14 are population-based algorithms. According to these algorithms, we generate random initial population in the first step. In the next step, we apply some operators to modify the initial population for next generation. We repeatedly use these operators until we reach the global optimum. When using Evolutionary Algorithms (EAs) for an MSA, an initial generation is generated by random manner, and then, the steps of an EA are applied to improve the similarities among the sequences. There are some evolutionary computations for MSA.15–19 There are some other genetic algorithm (GA)-based methods for MSA, such as SAGA, 19 GA-ACO, 20 MSA-EC, 21 MSA-GA, 22 RBT-GA, 23 GAPAM, 24 VDGA, 25 and MOMSA. 26 We define methodology of some algorithm to solve an MSA problem based on GA. In SAGA, the initial generation is generated randomly. According to SAGA, 22 different operators are used to gradually improve the fitness of MSA. But the problem behind SAGA is time complexity due to repeated use of fitness function. RBT-GA is also a GA-based method, combined with the rubber band technique (RBT), to find optimal protein sequence alignments. 27 RBT 28 is an iterative algorithm for sequence alignment using a DP table. The authors 26 solved 56 problems from reference sets 1, 2, 3, 4, and 5 of the benchmark Bali base 2.0 dataset and Bali base 3.0 dataset. The drawbacks of these evolutionary methods are also local optima due to poor diversity of the solutions.
Motivation and contributions
In the domain of biology, MSA is the most crucial to solve numerous standard problems such as structure prediction and phylogenetic property. According to the open literature, the MSA is still an open-challenging problem. Hence, we motivate to solve an MSA problem using the improved version of biogeography-based optimization (BBO). However, this paper achieves the following contributions.
We first proposed a method to improve migration operator in BBO and then used it in MSA for maintaining diversity of the solutions. The results obtained in experimental analysis are better in terms of time factor. In addition, we provide a comparison table, which claims that our method is better than the existing competitive solutions in terms of matching score.
Biogeography-Based Optimization
BBO 29 was designed by emigration and immigration of species from one habitat to another. In the BBO algorithm, candidate solutions are called habitats (or islands). Each feature in a solution represented by a habitat is called a suitability index variable (SIV), while the goodness of a habitat is measured by the habitat suitability index (HSI). Habitats with a high HSI can support more species, whereas low HSI habitats support only a few species. Poor habitats can improve their HSI by accepting new features from more attractive habitats in the evolution process.
In BBO, there are two main operators: migration and mutation. The migration operator is a probabilistic operator that can randomly modify SIVs based on the immigration rate λ

Main procedure of BBO

In BBO, the migration operator is a probabilistic operator that is used to randomly adjust each habitat 
Migration operator


Mutation operator

Proposed Method
Habitat representation
In BBO, each solution is represented as habitat.

In the initialization state, first put the gap in our given MSA randomly. The initial solution is given in Figure 1.
Initial solution.
Binary encoding scheme: In the encoding scheme, put 1 in the position of gap and put 0 in the position of protein sequences. Figure 2 shows an encoding of initial solution.
Encoding scheme.
After that, we are taking decimal value of this binary encoded value from bottom to top of each column. Hence, habitat representation of this solution is
Fitness function
The sum of pair is used to measure fitness of MSAs. Here, each column in an alignment is scored by summing the product of the scores of each pair of symbols. The score of the entire alignment is then summed over all column scores by using (3.2.1) and (3.2.2).

Here, 
Here,
New solution generation
In this process, two types of operators are used, one is migration and the other is mutation. To improve the solution, low HSI solution accepts the species from the high HSI solution. The entire process is called as migration.
Migration
Migration is used to diversify the solution space or to explore the solution search space, whereas mutation intensifies the solution search space. In each iteration, we are applying migration and mutation operators to the habitats. In the migration process, we share the feature of high HSI habitat to low HSI to improve the solution quality. This operator is very effective, and the resultant habitat is much more different from the actual habitat. We chose two habitats according to immigration and emigration rates. Afterward, one index was chosen randomly in emigration habitat, and this SIV/element goes to the same position of immigration habitat. This process is presented in Figure 3.
Graphical representation of migration process.
Mutation
Main procedure of IBBOMSA

Improved migration operator

Mutation operator


Graphical representation of mutation process.
Test Dataset
We have tested a large number of datasets from Bali base benchmark database to check the quality of our approach. Bali base version 1.0 30 contains 142 reference alignments, which keeps more than 1000sequences. Bali base version 2.0 31 is an extended version of Bali base version 1.0. Bali base version 2.0 contains 167 reference alignments, which keeps more than 2100sequences. Bali base version 2.0 contains eight reference sets. Each reference set keeps different types of sequences. Reference set 1 contains a small number of equidistance sequences. Reference set 2 contains totally different or unrelated sequence. Reference set 3 contains a pair of divergent subfamilies. Reference set 4 contains long terminal extension sequence. Reference set 5 contains large internal insertions and deletions. Finally, reference sets 6-8 contain test case problems where the sequences are repeated and the domains are inverted. Bali score is a score that measures the quality of algorithm. Bali score compares between manual alignment sequence (which is available on Bali base version 2.0) and alignment (which comes from some existence method). Range of Bali score is 0-1. If the manual alignment file and our output file are the same, then the score is 1. If the manual alignment file and our output file are totally different, then the score is 0. It gives the value between 0 and 1 according to similarity between Bali base manually alignment file and our output file.
Experimental Analyses
In this section, first, we compare IBBOMSA with the recently proposed MSA algorithms based on evolutionary algorithms, including VDGA, 23 GAPAM, 22 and MOMSA 24 to prove its dominance. After that, we also compare the performance of IBBOMSA with many well-liked aligners. In this paper, IBBOMSA is coded in C language and implemented in the personal computer in Linux platform.
Effect of improved operator in BBO
The BBO algorithm was invented for immigration and emigration of species between habitats in multidimensional search space. Each habitat represents a solution. In traditional BBO, migration features of good solution appear in poor solution as a new feature while still remaining in good solution. Since this feature may exist in several number of solutions, this may increase the exploitation capability and decrease the diversity of search space. An improved migration with in updated feature appears in poor solution, where updated features come from our proposed migration operator. We used one scaling function for maintaining the exploration (diversity) and exploitation capability. But we have to use this scaling function in a proper way to maintain diversity and exploitation capability. If Performance of improved BBO and some existing methods per generation with respect to reference set 1. (A) Performance of proposed method and other existing methods with respect to 1ped Data. (B) Performance of proposed method and other existing methods with respect to 1amk Data. (C) Performance of proposed method and other existing methods with respect to 1fieA Data. (D) Performance of proposed method and other existing methods with respect to 1ldg Data. Performance of improved BBO and some existing methods per generation with respect to reference set 2. (A) Performance of proposed method and other existing methods with respect to 1csy Data. (B) Performance of proposed method and other existing methods with respect to 1cpt Data. (C) Performance of proposed method and other existing methods with respect to 1havA Data. (D) Performance of proposed method and other existing methods with respect to 1sbp Data.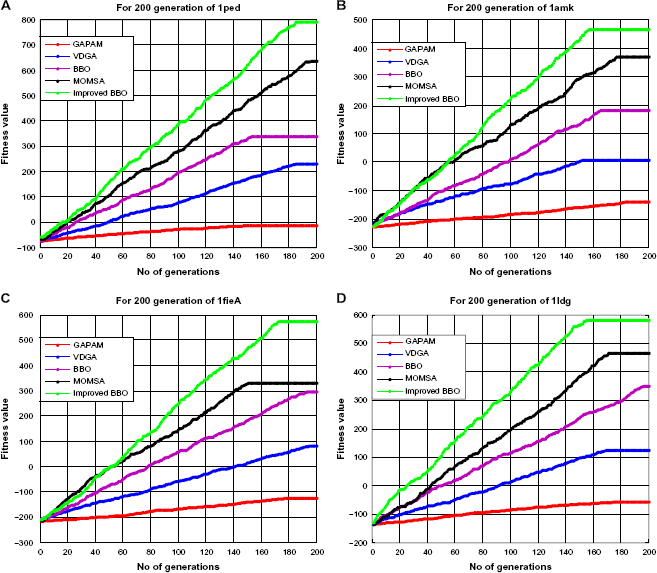

Experimental results and analysis
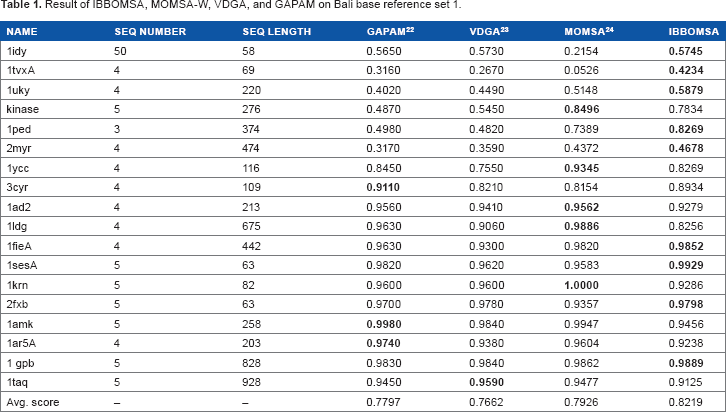
Result of IBBOMSA, MOMSA-W, VDGA, and GAPAM on Bali base reference set 1.
Result of IBBOMSA, MOMSA-W, VDGA, and GAPAM on Bali base reference set 2.

Result of IBBOMSA, MOMSA-W, VDGA, and GAPAM on Bali base reference set 3.

Result of IBBOMSA, MoMSA-W, VDGA, and GAPAM on Bali base reference set 4.

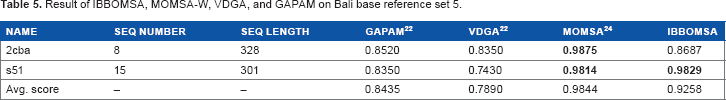
Result of IBBOMSA, MoMSA-W, VDGA, and GAPAM on Bali base reference set 5.
Comparison of IBBOMSA with MOMSA
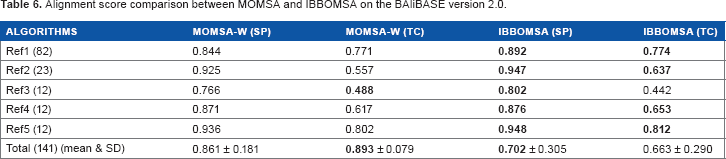
Alignment score comparison between MOMSA and IBBOMSA on the BAliBASE version 2.0.
Alignment score comparison between MOMSA and IBBOMSA on the BAliBASE version 3.0

Comparison of IBBOMSA with the state-of-the-art alignment algorithms
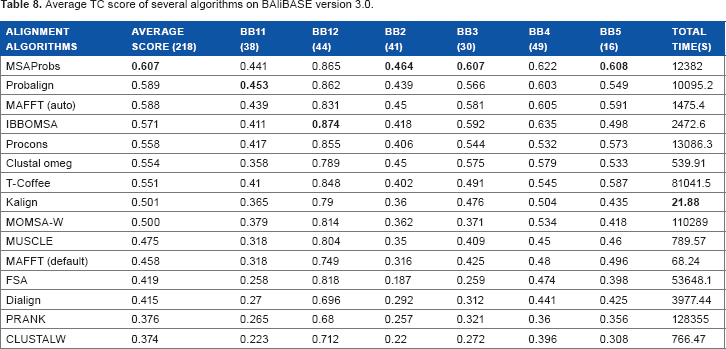
Average TC score of several algorithms on BAliBASE version 3.0.
Conclusions
In this paper, we have proposed an improved BBO algorithm for solving MSA. We design a new migration operator to maintain exploration and exploitation. However, we have to use scaling function carefully. We compared the new algorithm with the existing BBO algorithm. It shows that the new algorithm is superior to the existing BBO or at least competitive. To test our present approach, we considered a good number of benchmark datasets from Bali base 2.0, so as to cover all the test sets of MOMSA. Therefore, the corresponding Bali score of this solution was used to compare with other methods, as they used Bali score as their measure of the quality/accuracy of the
MSA. The experimental results proved that the proposed BBO performed better for most of the test cases. Since the solution of the proposed method was not best for some test cases, but it is close to the best. The proposed method performed better than the others because of its improved migration operator to help maintain diversity of search space. After the experimental analysis, we can say that the proposed method can effectively solve an MSA problem.
Author Contributions
Conceived and designed the experiments: RKY. Analyzed the data: RKY. Wrote the first draft of the manuscript: RKY. Contributed to the writing of the manuscript: RKY, HB. Agree with manuscript results and conclusions: RKY, HB. Jointly developed the structure and arguments for the paper: RKY, HB. Made critical revisions and approved final version: RKY, HB. Both authors reviewed and approved of the final manuscript.