
Abstract
We present EvalMSA, a software tool for evaluating and detecting outliers in multiple sequence alignments (MSAs). This tool allows the identification of divergent sequences in MSAs by scoring the contribution of each row in the alignment to its quality using a sum-of-pair-based method and additional analyses. Our main goal is to provide users with objective data in order to take informed decisions about the relevance and/or pertinence of including/retaining a particular sequence in an MSA. EvalMSA is written in standard Perl and also uses some routines from the statistical language R. Therefore, it is necessary to install the R-base package in order to get full functionality. Binary packages are freely available from https://sourceforge.net/projects/evalmsa/ for Linux and Windows.
Introduction
Multiple sequence alignments (MSAs) are essential instruments for prediction of protein structure and function, phylogenetic inference, and other tasks related to sequence analysis. 1 Data in MSAs are usually represented in a matrix form, such that the elements in the same column are homologous, occupy the same place in the genome/protein, and usually have a similar function in the protein structure. Because structure and function may diverge through time as a result of evolutionary processes, making such alignments becomes increasingly difficult when MSAs include sequences sharing a last common ancestor (LCA) very distant in time. 2 Sequences in an MSA whose last common ancestor is much more evolutionarily distant than that of the remaining sequences in the alignment can be denoted as divergent sequences. This scenario may also arise as a result of horizontal gene transfer processes and also through errors and mistakes during the different phases leading to an MSA. The methods used for aligning sequences try to maximize the number of matching residues by using a scoring scheme that penalizes mismatches and lack of homologous residues. To this end, gaps are introduced in the alignment. Usually, the more divergent the sequences, the more gaps are introduced to make homologous sequence elements match.3–5
Gaps in an MSA can result from the acquisition or loss of biological information represented by nucleotide or amino acid residues. Occasionally, it is difficult to identify the real cause between these two possibilities, but in any case, in order to maintain the homology in not fully conserved areas, it is necessary to insert gaps. In addition, current methods for constructing MSAs are not perfect and often introduce some errors and noise in the alignments.6,7 These errors become more frequent as more divergent sequences are included in an MSA, and they can seriously affect subsequent analyses.8,9
Filtering the alignments through the removal of positions (columns) of dubious homology is a common practice to increase the phylogenetic signal in the MSA and remove noise. 10 Most methods used in phylogenetic inference assume that sites evolve in an independent manner from each other. Thus, removing some columns is expected to enhance the signal-to-noise ratio in the MSA. The columns remaining after filtering are used to construct the phylogeny although evidence rejecting site independence has been published. 11 In consequence, several authors have proposed improving MSAs through the elimination of columns corresponding to weakly conserved or very divergent regions.10,12,13 Several software tools use this approximation. The popular gBlocks is based on searching for regions with contiguous conserved positions, with a minimum number of gaps and highly conserved flanking positions. 12 Other tools, such as T-Coffee, estimate the alignment confidence by a progressive approach. 14 GUIDANCE also calculates a confidence score that measures the robustness of the guide tree used for constructing the alignment. 15 Despite the popularity of all these tools, some authors indicate that current filtering methods still lead to inaccurate phylogenetic trees and new filtering methods and algorithms are needed. 16
More recently, a different approach has been taken. Sometimes, a divergent sequence is included in a group of conserved or close (in evolutionary terms) sequences. This inclusion can alter the whole alignment, and in that case, gaps must be introduced to maintain homologous and conserved areas. In many cases, gaps are inserted erroneously, and they not only become uninformative but can also decrease the quality of the global alignment. 17 The distortion introduced in the alignment affects not only a few columns but also almost the entire MSA. In these situations, and in order to improve the alignment quality, it is necessary to decide whether divergent sequences should be removed from the MSA. A similar question can appear when working with a large number of sequences obtained from public databases, and we must decide which of them should be included in the final analyses. We may accidentally include nonhomologous, wrongly identified, or even reversed sequences. All these situations can lead to an important loss of quality in the data and in a waste of time until we realize of these errors. In these cases, we should identify those divergent sequences as if they were outliers from the rest of the data. Outliers are patterns of data that do not match with the majority of the patterns in a dataset. The identification of outliers has been studied in the statistics community for a long time, and it is an important area in fields such as image processing, fraud detection, and medical anomaly detection. 18 Divergent sequences can act as outliers altering the results of posterior analyses and leading to erroneous conclusions.
OD-seq, a recently published tool, also identifies divergent sequences in an MSA by finding those cases with an anomalous average distance compared to the remaining sequences. 19 OD-seq computes all possible pairwise distances, counting the number of positions with gaps in one sequence and not in the other. Although this is a valid starting point, we consider that more information is needed to make a proper revision of the alignment.
We have developed a software tool to help users in the supervision, comparison, and decision-making tasks after obtaining an MSA. The principal goal of EvalMSA is to provide an objective view of the influence of each sequence on the quality of the MSA. We implement a novel method for detecting the contribution of each individual sequence to the insertion of gaps in the alignment, along with other classic methods such as sum-of-pairs (SP), and provide a statistical evaluation of the improvement in alignment quality if the offending sequences are removed from the MSA.
Methodology
EvalMSA performs different analyses in order to find divergent sequences or outliers in an
MSA. First, the program analyzes the original length of the sequences used to construct the
alignment. Then, EvalMSA calculates the contribution of each sequence to the alignment
quality to assess its relative importance in the MSA and assigns a weight to each sequence.
Finally, the tool evaluates the contribution of each sequence to introduce gaps in the
remaining sequences of the alignment by computing a parameter denoted
Preanalysis
Basic statistics on sequence length can readily detect the presence of wrong/misaligned sequences in an MSA, possibly as the result of unnoticed errors at selecting, downloading, or introducing sequence data into the alignment. In a preliminary analysis, EvalMSA reports some parameters related to the original sequence lengths, such as mean, median, standard deviation, quartiles, and outliers. Typical errors, such as including a single gene into an alignment of complete genomes, can be identified in this way.
Gappiness value
A truly divergent sequence, evolutionarily very distant from the rest, can introduce large gaps in a multiple alignment. Hence, we need to identify those sequences that introduce more gaps in the other sequences. To look for these sequences, we first identify the columns with more gaps than residues in that position. The method is independent of the type of residue (amino acid or nucleotide). For each sequence (row), we count the number of columns having a residue when most of the sequences have a gap. By calculating the number of gaps that each sequence generates in the rest, we can rank them by gap-generation capacity.
To evaluate the capacity of each sequence to introduce gaps in the alignment, a gappiness
value (
For a given sequence, the
Given an alignment with
For each column in the alignment, a
Then: 
An example of the application of this expression and more details can be found in the Supplementary files.
Evaluating the original alignment score and defining the gap penalty
For each alignment, the program calculates a score that will be a quality measure. To calculate this score, we use the SP method. The SP score takes into account all the pairwise information in the alignment and is one of the most popular, simple methods used for scoring an MSA. Using a scoring matrix in conjunction with the SP method allows us to score each sequence based on its similarity and mismatches with the rest of the sequences. A more detailed explanation about how the SP method is implemented in EvalMSA is provided in the Supplementary files.
This system can be used for assessing amino acid or nucleotide alignments through an appropriate scoring matrix. Scoring matrices typically do not include gap penalty values. One of the main problems with the SP method is the assessment of gap penalties. During the alignment process, inserting a gap in a sequence is always penalized. This penalty depends on how evolutionarily close the sequences are and on whether it corresponds to a gap opening or a gap extension. In our case, we will penalize all the gaps with the same value, but this can be changed easily.
Sometimes, the alignment process generates large gaps due to errors or the use of
inappropriate or very divergent sequences. These often lead to a serious decrease in the
quality of the alignment. We assume that, during the evolutionary process, losing or
gaining a residue is less likely than the substitution of an amino acid or nucleotide by
another. A gap in just one sequence means lack of homology (due to insertions or
deletions) to the rest of the sequences and represents a loss of information for
subsequent analyses. We account for mismatches and for gaps when scoring sequences, but we
want to maximize the gap effect and, in consequence, we penalize more those sequences with
more gaps. Hence, in our analysis, a gap will be penalized more than a mismatch and gap
penalties receive a lower value in the scoring matrix than perfect matches or mismatches.
For any set of values defined in the scoring matrix, we approximately obtain a normal
distribution (Fig. 1). The gap penalty
is defined as: Gap penalty value. Using all the values contained in the scoring matrix, we
obtained the distribution represented above. A value lower than the rest in the
scoring matrix is valued as a gap penalty.

If the alignment contains symbols that are not defined in the scoring matrix, such as *,
?, and X, they will be assigned the
Evaluating the weight of each sequence on the alignment quality
For each sequence in the MSA, we evaluate its weight (or influence) on the alignment quality. For this, each sequence is scored with a method derived from SP.
Given 
where 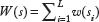
The
Benchmarking
MSAs used to benchmark the program.

Implementation
The application was implemented in Perl, with some modules imported from BioPerl. 23 The results are plotted with the R language, so it is necessary to install the R-base code in order to obtain full functionality of EvalMSA. Complete information about the algorithms and methods used to calculate the numerical values reported by EvalMSA can be found in the Supplementary files. We also provide the alignments used in the benchmarking as an example of use as well as the results of the analyses in the Supplementary files.
Results and Discussion
We benchmarked our application with six different alignments. Table 1 summarizes the different MSAs used to benchmark the program derived from the Pfam database. To describe the output of the program in detail, we will explore the results for alignment 5 in Table 1. To construct it, we took the seed alignment of the Pfam protein family Linocin M18 (PF04454). Then, we added three protein sequences from families belonging to the same Pfam clan (CL0373) to the seed alignment and we realigned it. We run EvalMSA with both alignments and compared the results. The program was run with default parameters in all the cases (default gap penalty, default gap threshold, the substitution matrix blosum62 for amino acid alignments, and DNA2 for the nucleotide one).
The returned results can be easily interpreted. Figure 2A (left) shows an example of the preanalysis output, in which a usual
sequence length distribution is shown. All the sequences have a similar length (only 30 bp
of difference between the shortest and the longest sequences). In the right part of Figure 2A, it is possible to identify a pair
of sequences that radically differ from the others in terms of sequence length. The weight
distribution, as shown in Figure 2B, is
also an intuitive way of evaluating the contribution of each sequence to the alignment
quality. The left histogram shows that most sequences have a similar weight. The magenta
line identifies the sequence with the highest gappiness value, and the green line identifies
the sequence with the largest number of gaps. This sequence contributes markedly less than
the rest to the alignment quality. The weight of the sequence identified as introducing more
gaps (magenta line) does not differ much from the rest of the sequences. On the other hand,
the right histogram shows that four sequences had less weight values than the rest. The
sequences marked as having more gaps and the one provoking more gaps are in the left part of
the histogram. The same information is represented in Figure 2C but after normalizing the weight scores and sorting
them. In this way, it is easier to appreciate the differences among weighting scores. In the
left plot, most of the sequences have a high score and only one of them has a low weight. In
the right plot, we can see that at least four sequences have a weight value lower than the
rest. Three of these sequences correspond to the artificial outliers that we added to obtain
the MSA. Finally, the distributions of gappiness values are shown in Figure 2D. Again, by comparing the right and the left plots,
we can observe that two of the outliers have higher gappiness values than the rest. Default output. (A) Preanalysis boxplot showing the original sequence length
distribution. (B) Weight score histogram, highlighting the sequences with the highest
number of gaps (green line) and with the largest gappiness value (magenta line). (C)
Normalized weight score distribution. Sequence index refers to the list of sequences
listed by weight. (D) Gappiness values. Sequence index refers to the list of sequences
listed by summary of the results obtained after running the program with dataset 5
aligned with MUSCLE (see Table
1).

The plots shown in Figure 2 correspond to the standard output of the tool. The program also writes a Comma Separated Values (CSV) file with all the values calculated associated with the sequence names, so additional analyses can be made by the user. Table 2 shows the CSV file obtained from the above example, in which we can see that the sequence with less weight value corresponds to outlier 2, while the two sequences with the highest gappiness values are outliers 3 and 1. These results mean that outliers 3 and 1 are the sequences that introduce more gaps in the rest of the sequences. So, probably, their presence is altering the whole alignment. The user can identify them easily as possible outliers. On the other hand, the gappiness value for outlier 2 is really low, so it does not introduce many gaps in the rest of sequences. However, the weight of this sequence is the lowest one so the number of gaps and mismatches is higher than for the rest of the sequences. Hence, outlier 2 sequence should also be taken into account by the user as a possible outlier. In addition to these files, a plain-text file with a short summary of the divergent sequences identified is created.
We have not observed remarkable differences between alignments obtained with different algorithms, such as those implemented in MUSCLE and ClustalW. The outliers were identified correctly in both cases. Moreover, we used the Expresso algorithm 21 to construct alignments taking structural information into account. Again, no remarkable differences were found in the results. So, our program works independently from the algorithm used to align the data. In addition, we have checked that our program correctly identifies outliers from other data sources such as GenBank sequences. In Supplementary file 2, we have included an alignment of 13 mitochondrial genomes from primates and one mitochondrial genome from a reptile. EvalMSA correctly identifies the reptile sequence as the one with the least weight as well as the highest gappiness value.
Figure 3 presents the results of a
benchmark test showing the execution time of the program when run in a personal computer
(Intel Core i7-4770 CPU 3.40 GHz, 16 GB RAM, 1 TB HDD) for different MSAs. We tested the
program with an MSA of 415 virus and 20 bacterial genomes. The execution time is fast with
short (virus) genomes, but it slows down as the number of sequences in the MSA increases.
The most time-consuming part of the program is the weight computation. It relies on a nested
loop that goes over all the positions for each sequence. So, the time complexity for the
program is Execution time of the program with different alignment sizes. (A) Execution
times for MSA with different numbers of sequences (sequence length 10 kbp, viral
genome). (B) Execution times for MSA with different numbers of sequences (sequence
length 4.4 Mbp, bacterial genomes).
By default, EvalMSA calculates a gap penalty that depends on the substitution matrix used
to score the alignment. This value, as well as the substitution matrix and the gap threshold
used to calculate the
Although a similar tool was recently published,
18
the software presented here provides a wider
perspective on sequence divergence and can be used complementarily to OD-seq. Similarly to
OD-seq, EvalMSA also penalizes gap occurrence, but it takes into account mismatches,
To compare both programs, we analyzed alignment 5 in Table 1 with OD-seq. This program only identified as
outliers the introduced sequences 1 and 3 and maintained outlier 2 in the core alignment.
The identification of outliers by OD-seq is based on the number of gaps between sequences,
but EvalMSA also penalizes mismatches and takes into account gappiness values. Hence, our
program can identify the different outliers and the cause of their divergence from the rest
of the sequences (
Evaluating alignments is not a trivial or obsolete problem. The decision of including or not a sequence in a multiple alignment and subsequent analyses is ultimately made by the investigators. It is important to have a method that allows them to evaluate MSAs with a unique methodology and criterion, so that decisions are based on objective data. Our software tool serves as an objective data provider so that investigators can make decisions based on an objective, easily reproducible method besides their own experience and criterion. Finally, EvalMSA was programmed using Perl and R, so it is portable to almost any platform.
Author Contributions
Conceived and designed the experiments: FG-C, AC-O. Analyzed the data: AC-O. Wrote the first draft of the article: AC-O. Contributed to the writing of the article: FG-C, AC-O. Agreed the article results and conclusions: FG-C, AC-O. Jointly developed the structure and arguments for the article: FG-C, AC-O. Made critical revisions and approved the final version: FG-C, AC-O. Both authors reviewed and approved the final article.
Supplementary Material
Supplementary File 1
Document file with additional details on the methods used in EvalMSA.
Supplementary File 2
Alignment files with and without outliers used to benchmark the program.
Footnotes
Acknowledgment
We thank Dr Garcia-Garcerá for his help and comments in testing EvalMSA during its development.