
Abstract
Plants are simultaneously subjected to a variety of stress conditions in the field and are known to combat the hostile conditions by up/downregulating number of genes. There exists a significant level of cross-talk between different stress responses in plants. In this study, we predict the interacting pairs of transcription factors that regulate the multiple abiotic stress-responsive genes in the plant
Introduction
Our understanding of the plant adaptation to various kinds of stress conditions at the molecular level has increased considerably over the years. Various kinds of abiotic stresses limit crop productivity in natural conditions. Abiotic stress negatively influences survival, biomass production and accumulation, and grain yield of most crops. Different crop ecosystems are affected by several abiotic stress factors, and to a differential extent. Insights into the responses elicited in plants by different types of stress have been obtained by studying the genes regulated (up/down) during these stress conditions. 1 5 It has been documented that there exists a significant cross-talk between the signal transduction pathways activated during different abiotic stress conditions. 6 12 There have been several attempts to explore and understand this cross-talk between signaling pathways using rice as a model organism. 11
The current study aims at studying the transcription factors (TFs) that are responsive to multiple abiotic stress conditions in
The interaction between different TFs can also be exploited to generate diversity in controlling the gene expression.15,16 The diverse set of eukaryotic genes are regulated by small kinds of TFs and it is the different combination of these protein factors which regulate the expression of different genes.15,17,18 Physical interactions between the multiple TFs bound upstream of every gene may dictate the specific combination of conditions under which the gene will be expressed. Also, in 2011, the role of TFs in combating stress conditions by generating drought-resistant crops was stressed by manipulating the expression of drought-responsive genes with the help of TF DREB/CBF.16,19 There have been many other studies in the field, emphasizing the role of different TFs in regulating the stress responses in plants.16,20,21
Therefore, studying physical interactions between the pair of TFs, known to bind the multiple abiotic stress-upregulated genes, will further prove useful to understand how combinatorial regulation of gene expression is achieved in order to combat multiple stresses.
Protein-protein interactions constitute the “interactome” of the cell and dictate majority of cellular processes and are known to regulate responses of organisms to varied environments including stress conditions. 20 There are excellent biochemical/experimental techniques like yeast two-hybrid, co-immunoprecipitation, which aim to identify the interacting pairs of proteins, but they are usually time-consuming. Further, not all pairs of proteins can be tested for their interactions using these methods. Therefore, it would be interesting to computationally predict the interacting pairs of proteins that might be regulating the multiple abiotic stress-responsive genes, followed by their detailed experimental validations.
To accomplish this, we selected
Methods
Master Genes Identification
An
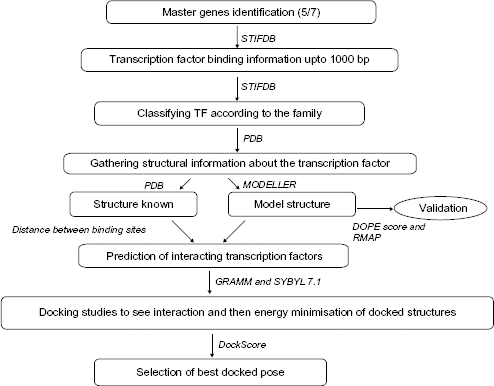
Workflow describing the method and tools/techniques adopted.
Information on TFBS, 1000 bp upstream of the master genes, was also obtained using STIFDB (TFBS with Z-score >1.5 were used for further analysis).
TFBS on Master Genes
A pair of TF was expected to exhibit a combinatorial control of the master gene through physical interaction, if their TFBS were placed less than 50 bp apart. The 50 bp cut-off is used in some of the earlier studies to examine the formation of
Molecular Modeling and Validation
The structural information for all the putative interacting pairs was gathered using PDB. 26 In the case of TF with unknown structures, comparative modeling technique was used employing MODELLER 9v7. 27 The template used to perform modeling was selected based on sequence homology and atomic resolution of the structure.
The five low-energy modeled structures obtained based on the Modeller's DOPE score 28 were further validated by performing Ramachandran map analysis (model structure with maximum number of amino acids in allowed regions) using PROCHECK server. 29
Molecular Docking
Docking was performed using GRAMM 30 and 10 docked poses were obtained for all the putative interacting pairs. The docked model was further subjected to energy minimization using the SYBYL software package (Version 7.1) (Tripos Associates Inc., St. Louis, MO). Tripos force field was used for minimization (100 iterations with electrostatics off) to obtain final negative energy (kcal/mol) of the complex structure.
Scoring Docked Poses
The poses obtained subsequent to docking were scored using our scoring scheme DockScore in order to identify the optimal interactions between the putative interacting TFs. The top-ranking pose was selected as the near-native complex.
Results
Identification of Master Genes and Their TFBS
Fifteen master genes, expressed in five or more stress conditions, were identified from STIFDB (Table 1). TFBS data, 1000 bp upstream of these genes, were also collected from STIFDB to identify the regulators of master genes (Supplementary Table 1 and Supplementary Table 2). The schematic exemplifies the nature and position of TFBS for one of the master gene (AT4G27410, Supplementary Fig. 1). The distances between TFBS were analyzed and the frequency matrix was constructed for the number of pairs of TFBS on master genes located <50 nucleotides apart (Table 2). Four pairs of TFs MYB-bHLH, MYB-ARF, HSF-WRKY, and bHLH-bZIP, having TFs from six different families (the highest frequency in the matrix), were selected (Table 2) and designated as putative interacting pairs. These four putative interacting pairs were further tested for the presence and the nature of interactions.
Master genes with stress conditions they are upregulated in Identification of 15 master genes selected out of 2629 abiotic stress responsive genes in

Frequency matrix for interactions between transcription factors. The transcription factors predicted to bind 15 master genes belong to 9 classes. For each of the master genes if the distance between the two successive binding sites is ≤50, then it is given the score of 1. This matrix records the score for every 45 possible pairs of transcription factors (frequency matrix). The score marked with asterisk corresponds to the pair having maximum frequency and were named as “putative interacting.”

Interactions among TFs
TFs regulating master genes, as documented in STIFDB, belong to nine different families. Out of the above-mentioned six families of TFs, structures are available for two TFs (MYB and WRKY) and for rest of the four families, structures were modeled by comparative modeling techniques (Table 3). Out of five best models obtained for each of the TF (please see Methods for details), one model with least DOPE score and highest percentage of allowed regions in Ramachandran map was selected as the best (Fig. 2). The TFs, bHLH and bZIP, are known to exist as either homo or heterodimers in the cell. However, we modeled bHLH both as a homodimer using multi-chain modeling and single chain so as to study its interactions with different TFs, MYB and bZIP. As bZIP and bHLH are known to heterodimerize, 31 bZIP was modeled as single chain to study its interaction with bHLH TF. Among these four pairs of TFs studied, we also validated their interaction using the BioGrid database. bHLH is reported to form homodimer as well as it can heterodimerize with the bZIP TF32,33 In the BioGrid database, the interaction data for WRKY is still not curated; however, there is an earlier report that stated WRKY and HSF co-express during oxidative stress conditions. 34
Putative interacting pairs of transcription factors and their details of their structural data. The table highlights the PDB ID of the template used for modeling the transcription factor along with its percentage identity with the query and resolution of the template. It is appropriately listed, if the structure of a transcription factor is already deposited in PDB. For transcription factors, MYB and WRKY, the structures were there in PDB, whereas for bHLH, bZIP, ARF and HSF, the structures were modeled using comparative modeling.

PDB ID of crystal structure if known else of the template used for modeling.

Modeled structures of transcription factors using Modeller (9v7) (
Using these structures, interactions between the four pairs of TFs were studied with the help of molecular docking using GRAMM. Ten docked poses for each of the TF docking were generated which were further selected by implementing our scoring scheme DockScore.
TF Pairs Subjected to DockScore
After testing and assessing the performance of DockScore on the testing dataset comprising of 30 protein-protein complexes, 22 the four pairs of TFs identified as putative interacting pairs were subjected to DockScore in order to identify the docked pose with optimal interactions. The docked pose obtaining the highest score was selected as the best-docked pose (Fig. 3).

Docked posed pairs of transcription factors and their interface analysis.
The best-docked poses selected for the four pairs were further analyzed for the interface residues and their conservation using ConSurf. 35 The interface formed upon interaction is rich in conserved residues (marked in orange in Fig. 3).
For the TF pair MYB-bHLH, it is previously reported that the N-terminus region of bHLH is involved in interaction with MYB TF. 36 The docked pose we selected (for bHLH and MYB) also bears the interface residues at the N-terminus of the bHLH. The literature evidence also supports interaction between the TFs MYB-ARF and MYB is known to interact with C-terminus of ARF. 37 The docked pose we obtained for this pair of TFs possess interface residues at the C-terminus of ARF. bZIP and bHLH are reported to form high molecular weight complexes, suggesting functional relationship between the two. 31
Discussion
Plants respond to different stress conditions by either upregulating or downregulating the expression of some genes. The regulation of gene expression is a very vexed mechanism in eukaryotes and it is accomplished with the help of different TFs. The function of genes is highly orchestrated by the action of these protein factors. Therefore, to understand the details of gene regulation, studying interactions between different TFs will be of great value.
Also, as a response to different stress conditions, plants are known to upregulate some general as well as stress-specific genes. The present study deals with the genes elicited in response to multiple stress conditions. We observed that some genes were upregulated in multiple abiotic stress conditions and we named them as “master genes.” There were 15 master genes, identified in
Since accurate structure determination of macromolecular complexes are highly challenging, prediction of protein-protein interactions through molecular docking is highly appropriate. However, implementing molecular docking, for studying the interactions between a pair of proteins poses a challenge to identify the best-docked pose out of the pool of various poses suggested by the docking program GRAMM. For identifying the best-docked pose, we devised an objective scoring scheme, named DockScore, which takes into account several interface parameters and hence ranks the docked poses. The four pairs of TFs were subjected to DockScore and the best-docked pose was selected as the one with the highest DockScore.
In future, more TF pairs will be analyzed in detail to validate the interactions between them, even as recorded in STIFDB2 database. 38 Also, employing the similar approach, interactions between the TFs upregulating stress-specific genes and other multiple stress-responsive genes can be studied.
These kinds of studies will aim to provide detailed insights into the regulation of stress-responsive genes at the level of transcription. Also, the existence of interaction among protein factors regulating the responses of plant under stress conditions will provide an additional level of regulation on one hand, and will also lead to combinatorial diversity of regulatory complexes. With different combinations of these factors, regulation of diverse numbers of genes can be achieved. Therefore, studying physical interactions among TFs will provide useful insights into unraveling the basis of this combinatorial diversity in eukaryotes.
Conclusions
Plants are continuously exposed to a number of stress conditions in the fields and they have developed stress resistance or tolerance mechanisms since they are sessile in nature. They achieve this by up/downregulating some genes, which are termed as stress-responsive genes. We have studied transcriptional regulation of multiple stress-responsive genes in
Author Contributions
Conceived and designed the experiments: RS. Analyzed the data: SM. Wrote the first draft of the manuscript: SM. Contributed to the writing of the manuscript: RS. Agree with manuscript results and conclusions: SM, RS. Jointly developed the structure and arguments for the paper: SM, RS. Made critical revisions and approved final version: RS. Both authors reviewed and approved of the final manuscript.
Supplementary Data
Supplementary Table 1
Transcription factor binding site data (from STIFDB) for one of the master gene.
Supplementary Table 2
The TFBS information for rest 14 master genes. The URL provides 1000 bp upstream TFBS information for the respective master genes.
Supplementary Figure 1
Transcription factor binding site data (from STIFDB) for one of the master gene.
Footnotes
Acknowledgements
The authors thank NCBS (National Centre for Biological Sciences) for infrastructure and other facilities.