
Abstract
After iron, zinc is the most abundant essential trace metal. Intracellular zinc ([Zn]i) is maintained across a wide range of cells and species in a tight quota (100 to 500 μM) by a dynamic process of transport, intracellular vesicular storage, and binding to a large number of proteins (estimated at 3-10% of human proteome). As such, zinc is an integral component of numerous metalloenzymes, structural proteins, and transcription factors. It is generally assumed that a vanishingly small component of [Zn]i referred to as free or labile zinc, and operationally defined as the pool sensitive to chelation (by agents such as N, N, N', N'-tetrakis [2-pyridylmethyl] ethylenediamine [TPEN]) and capable of detection by a variety of chemical and genetic sensors, participates in signal transduction pathways. Zinc deficiencies, per se, can arise from acquired (malnutrition, alcoholism) or genetic (mutations in molecules affecting zinc homeostasis, the informative and first example being acrodermatitis enteropathica) factors or as a component of various diseases (e.g., sickle cell disease, cystic fibrosis, sepsis). Hypozincemia has profound effects on developing humans, and all facets of physiological function (neuronal, endocrine, immunological) are affected, although considerably less is known regarding cardiovascular pathophysiology. In this review, we provide an update on current knowledge of molecular and cellular aspects of zinc homeostasis and then focus on implications of zinc signaling in pulmonary endothelium as it relates to programmed cell death, altered contractility, and septic and aseptic injury to this segment of the lung.
Keywords
The role of zinc was first reported in 1869 when it was discovered to be important for the growth of Aspergillus niger.[1] Zinc was not recognized to be important for human life until 1963 when zinc deficiency was discovered as a major contributing factor in nutritional dwarfism syndrome and hypogonadism.[2] It is now well established that zinc is important for numerous cellular functions including cell differentiation[3] and division,[3,4] DNA synthesis,[4,5] RNA transcription,[4,5] and maintaining plasma membrane integrity.[6] Recent approaches using bioinformatics methods to mine existing protein databases indicate that approximately 10% of the human proteome is zinc dependent.[7] Zinc plays three major biological roles: a structural component of at least 3,000 proteins,[8] including transcription factors,[8] cytokines, and receptors;[8] a catalytic component of more than 300 enzymes[9] that regulate many cellular activities including DNA synthesis and maintaining membrane stability;[10–12] and a regulator of enzyme activity by acting as an activator or inhibitor ion.[10] Total intracellular zinc is maintained in a concentration range from 100 to 500 μM[13] across numerous cell types. Zinc is considered a trace metal; however, this is because more than 99% of intracellular zinc is protein bound. The concentration of labile [Zn]i is vanishingly small with estimates between 10−9 M[14,15] to 10 −12 M[16] and it is this fraction that may act as a second messenger in cell signaling[17,18] in a fashion well supported for other divalent cations such as calcium.
Zinc has been referred to as a “double edged sword”[19] as both zinc deficiency and zinc excess are associated with adverse effects on cell physiology.[11,20,25] Zinc deficiency stimulates inter-nucleosomal DNA fragmentation and apoptosis in intestinal,[26] neural,[27] respiratory epithelium,[28] and in systemic endothelium,[29] and high levels of zinc (> 250 μM) are associated with concentration dependent increases in cell death in cultured pulmonary[30,31] and cerebral[32] endothelia. In contrast, lower zinc concentrations (10 μM)[33,35] have been shown to inhibit cadmium-,[35] linoleic acid-,[33] and tumor necrosis factor-α (TNF-α)-[33] induced apoptosis in systemic endothelial cells. At the systemic level, the following occurred: labile [Zn]i levels were demonstrated to be affected by changes in fluid shear stress levels in mouse aorta and in human umbilical vein endothelial cells indicating that zinc dyshomeostasis in the systemic endothelium may contribute to the development and progression of cardiovascular diseases; and zinc supplementation was shown to reverse systemic inflammation and organ damage, with a positive effect on overall mortality in mouse model of sepsis.[35] Little is known about the signaling role of labile [Zn]i in the pulmonary endothelium in the context of lung diseases. In this review, we discuss the impact of zinc homeostasis and signaling, as well as its efficacy as a cyto-protectant in pathophysiological processes of pulmonary endothelial cell injury and death.
INTRACELLULAR ZINC HOMEOSTASIS
Intracellular zinc concentration is maintained by the coordinated activity of a large family of zinc transporters (ZnT and ZIP)[37] and zinc binding proteins such as metallothionein (MT)[37] (Fig. 1). Zinc transporters are encoded by one of two of the solute-linked carrier (SLC) gene families: SLC 30 (also known as zinc exporters or ZnT1-10);[38] and SLC39 (also known as zinc importers or ZIP1-14).[38] ZnT transporters reduce cytoplasmic zinc by promoting zinc efflux from cells or into intracellular vesicles, while ZIP transporters increase cytoplasmic zinc by promoting zinc influx from extracellular and, perhaps, from vesicular stores into cytoplasm.[39]
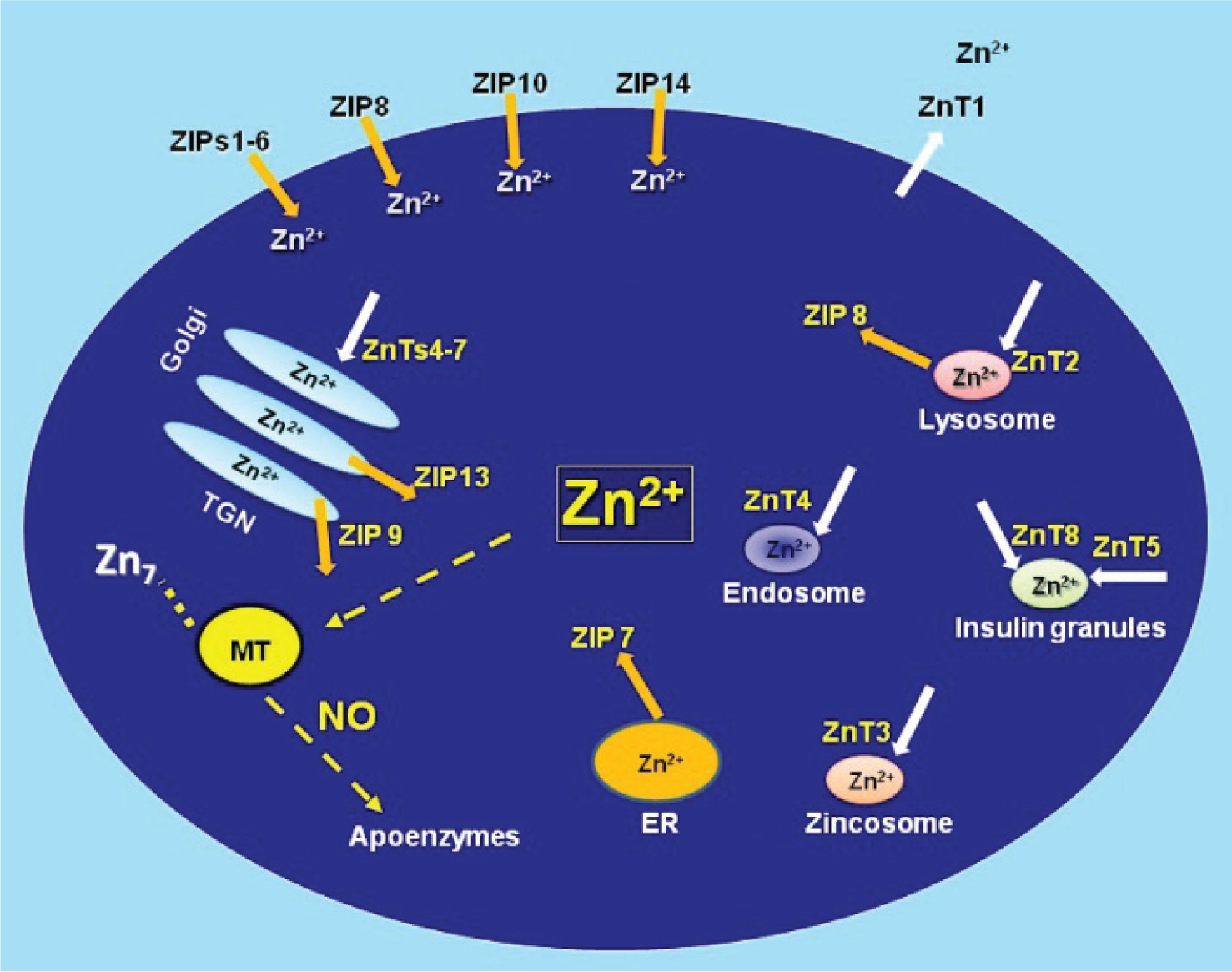
Regulators of intracellular zinc homeostasis. The labile pool of intracellular zinc is tightly controlled by zinc importers (ZIPs), zinc exporters (ZnTs), zinc storing vesicles, and zinc binding proteins such as metallothionein (MT). MT plays a critical role in zinc homeostasis acting as a buffer in the steady state while controlling the cellular distribution of transiently elevated zinc in response to perturbations and/or agonists such as nitric oxide (NO).[141] Modified figure from references.[74,142,143]
Metallothioneins
Metallothioneins (MT) are major zinc binding proteins that dynamically coordinate up to 7 mol Zn2+/mol MT via cysteine residues (approximately mol 30%).[40] MT is involved in the following: detoxification of heavy metals like mercury, cadmium, and alkylating cancer drugs;[41,42] scavenging free radicals;[41] and protection against DNA damage,[41] oxidative stress,[41] and apoptosis.[43] Mammals express at least four isoforms-MT-1, MT-2, MT-3, and MT-4. In humans, there are at least 16 MT genes located in chromosome 16 and most of them are associated with the MT-1 isoform.[44] MT-1 and MT-2 are expressed in many tissues and are particularly abundant in the liver, pancreas, intestine, and kidney.[45] MT-3 and MT-4 are minor isoforms with specific expression patterns in brain (MT-3) and stratified squamous epithelial cells (MT-4).[46] At the subcellular level, MT can be localized to a number of cellular compartments (i.e., mitochondria, cytosol, and nucleus)[47] as well as in the extracellular space.[48] The reduction potential of MT (less than −366mV[40]), makes it highly sensitive to physiological oxidants. We[49] and others[50–53] have shown that MT is sensitive to changes in cellular redox state and demonstrated that increases in reactive oxygen[54] or nitrogen[55,56] intermediates can oxidize or transnitrosate cysteines in its zinc sulfur clusters leading to liberation of zinc. As such, MT can be viewed as acting as a sensor and switch and connecting changes in cellular redox status with alterations in labile zinc (Fig. 1).
ZIPs
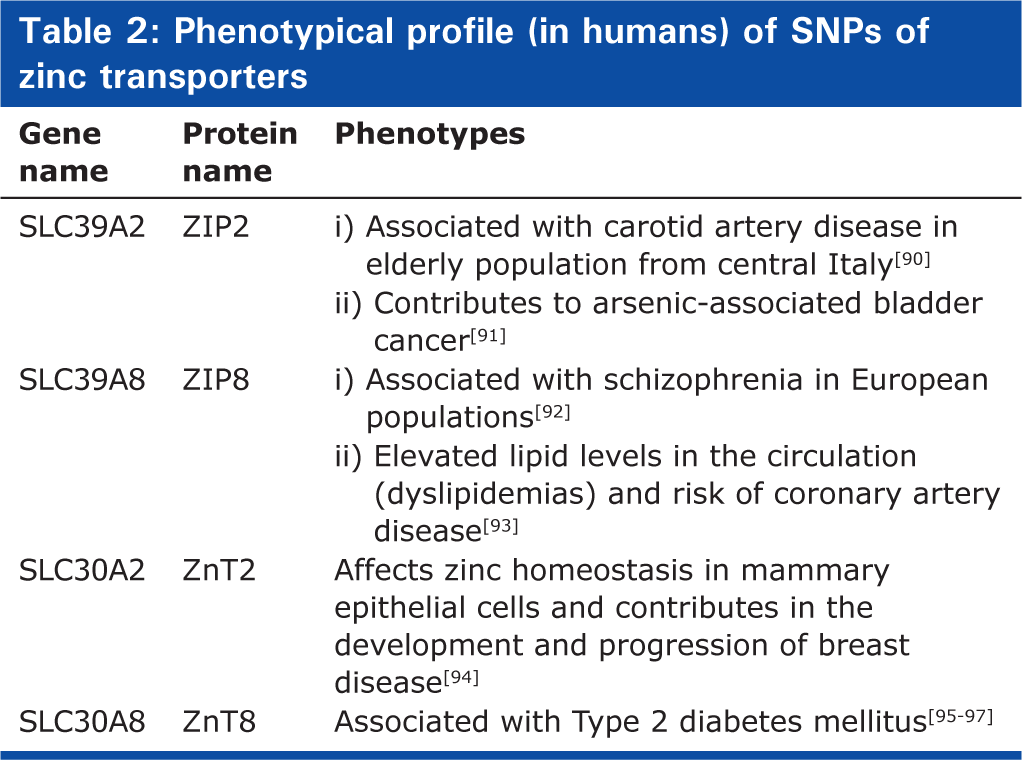
Fourteen ZIP family members have been reported in mammals.[57–59] The majority of ZIP family members are located on the plasma membrane[60–66] with the exception of ZIP7-8 and ZIP13 (Fig. 1) that are present in intracellular organelles. Gene knock-out technologies have provided valuable information regarding biological significance of the ZIP family members. Knockout (KO) mice lacking ZIP1, ZIP2, and ZIP3 are reported to have abnormal embryogenesis under zinc-limiting conditions.[62,67,68] ZIP4 KO mice embryos die during early development, whereas heterozygous mice exhibit a phenotype similar to acrodermatitis enteropathica (AE) secondary to impairment of intestinal absorption of zinc.[69–71] ZIP13 KO mice suffer from disorganization in hard connective tissue, including bone, teeth, skin, and eyes.[72] In humans, lack of ZIP13 is associated with spondylocheiro dysplasia, a form of Ehlers-Danlos syndrome.[72,73] Mice lacking ZIP14 have impaired G-protein coupled receptor (GPCR) signaling[74] and exhibit retarded growth and impaired gluconeogenesis. A summary of phenotype in genetically ablated or spontaneous mutants in various species (including humans) is provided in Table 1. Recent reports on association of single nucleotide polymorphisms of various zinc transporters with human disease are summarized in Table 2.
Phenotypical profile (in mouse, *human, **drosophila, ***sheep) of mutants of zinc transporters
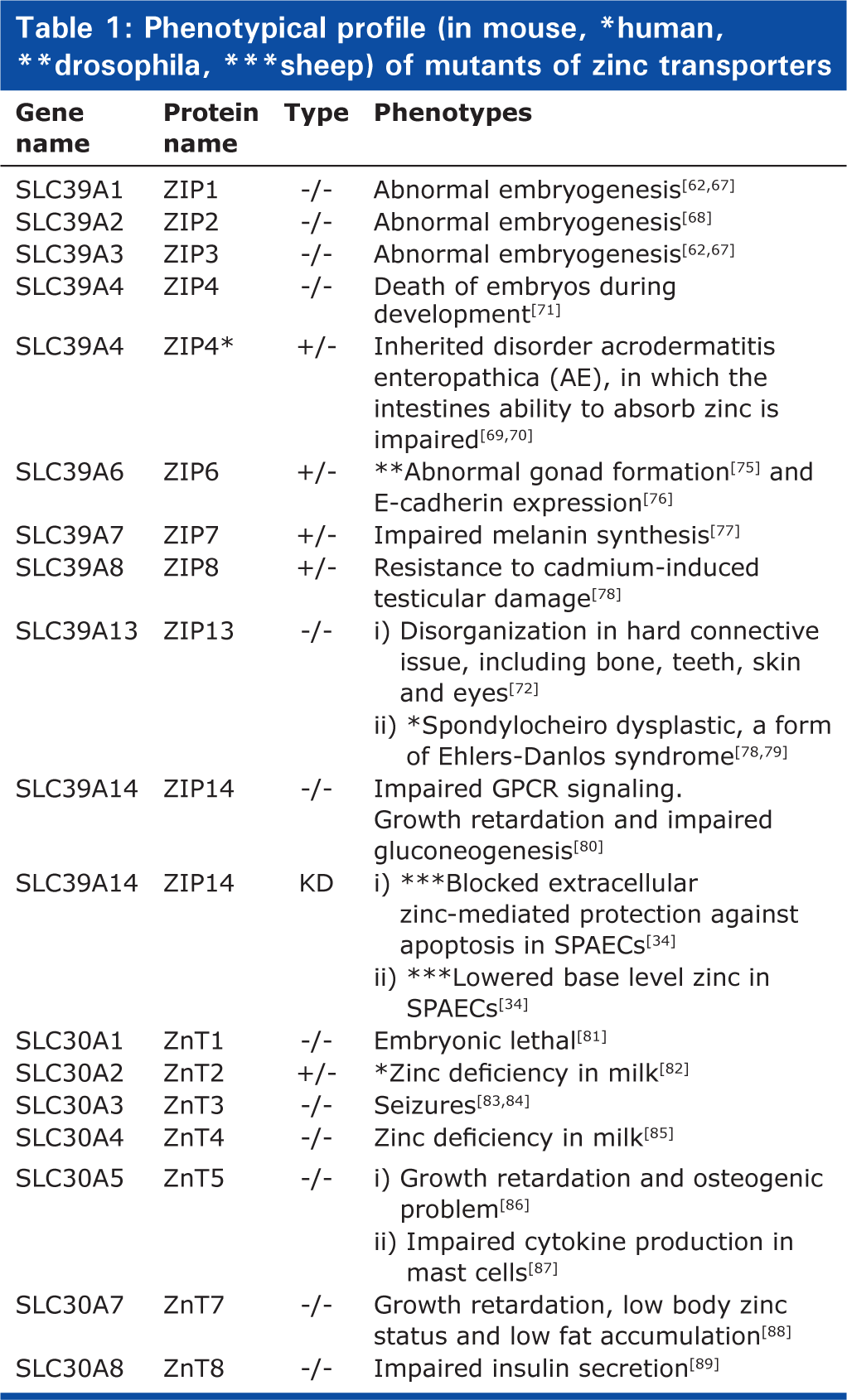
Phenotypical profile (in humans) of SNPs of zinc transporters
ZnTs
Ten ZnT family members have been reported in mammals.[59] Most ZnTs are located on intracellular organelles (i.e., golgi, endosomes, and endoplasmic reticulum)[98] (Fig. 1). ZnT 1 is the only ZnT exporter located at the plasma membrane, compatible with its role as the primary regulator of cellular zinc efflux.[99] ZnT1 knockout mice are embryonic lethal.[83] Disruption of the ZnT genes yields diverse phenotypes providing insight into the biologic function and specificity of the various family members. Mutations in ZnT2[82] and ZnT4[85]result in the production of zinc deficient milk in women and mice, respectively. ZnT3 knockout mice are prone to seizures.[83] Mice lacking in ZnT5 show growth retardation and osteogenic problems[86] and exhibit impaired cytokine production in mast cells.[87] Single-nucleotide polymorphism (SNPs) in ZnT8 are associated with type 2 diabetes in humans,[95] and deletion of the ZnT8 gene results in impaired insulin secretion in mice[89] (Table 1).
Manipulation of intracellular zinc levels have been shown to influence the expression and localization of zinc transporters[59] with reports of increased expression of members of ZIP family and decreased expression of ZnT family members in response to decreases in intracellular zinc/[100–104] Most of these studies have been performed in intestinal and respiratory epithelial or immune cells. While reported increases in ZIP1 and ZIP14 mRNA were shown to be normalized by dietary zinc supplementation in a mouse model of acute lung inflammation,[101] the mechanisms underlying the association between zinc homeostasis and lung disease remain largely unknown. ZIP6 was shown to play a role in blocking LPS-induced decreases in intracellular labile zinc and consequent maturation in mouse dendritic cells.[105] Zinc mediated cytoprotection against TNF-α-induced damage in human lung epithelial cells was shown to be dependent upon expression ZIP8.[38] We recently reported[34] the following in cultured sheep pulmonary artery endothelial cells (SPAECs): ZIP14 is sensitive to changes in intracellular labile zinc; and exogenous zinc mediated protection against LPS-induced apoptosis is dependent upon ZIP14.
ZINC HOMEOSTASIS IN THE PULMONARY ENDOTHELIUM
The Zalewski laboratory in Adelaide, Australia were the first to image labile zinc in the airway[12,106] and provide evidence that zinc chelation (via TPEN) enhanced hydrogen peroxide-induced caspase activation.[106] As reviewed by Troung Tran et al.,[4] intracellular zinc has also been shown to be important for ciliary function, wound healing (via re-epithelialization), and suppression of oxidative stress and apoptosis in the airway epithelium. Further evidence suggests that zinc deficiency sensitizes the lung to acute lung injury following alcohol induced epithelial dysfunction.[107] hyperoxia,[108] and polymicrobial sepsis.[36,109]
We have shown that zinc chelation (via TPEN) exacerbates LPS-induced apoptosis in pulmonary endothelium (SPAECs).[30] TPEN also reversed the protective effect of nitric oxide (NO) donors on LPS-induced apoptosis.[110] More recently, we reported that LPS induced time-dependent decreases in intracellular labile zinc (Fig. 2) in SPAECs using both live cell imaging and fluorescence-activated cell sorting (FACS) with the zinc-sensitive fluorophore, FluoZin-3 (Life Technologies, Grand Island, N.Y.).[111] We further verified the observed decrease in FluoZin-3 detectable zinc using a chimeric reporter encoding a zinc-sensitive metal-response element (MRE) fused to a luciferase gene.[111] The LPS-induced changes in labile zinc were accompanied by increases in ZIP14 mRNA. These effects were blocked by addition of exogenous zinc, as was LPS-induced apoptosis (increased caspase 3/7 activity and PS externalization).[111] In separate studies in SPAEC, siRNA knockdown of ZIP14 decreased basal levels of intracellular labile zinc and blocked zinc uptake (as determined by FluoZin-3), and abrogated zinc mediated protection against LPS-induced apoptosis (observed in WT and scrambled control).[34] Collectively, these data suggest that endogenous levels of labile zinc can modulate the sensitivity of pulmonary endothelium to the proapoptotic effects of LPS (Fig. 2) and implicate ZIP14 in affecting the ability of extracellular zinc to inhibit LPS-induced apoptosis in SPAEC (Fig. 3).
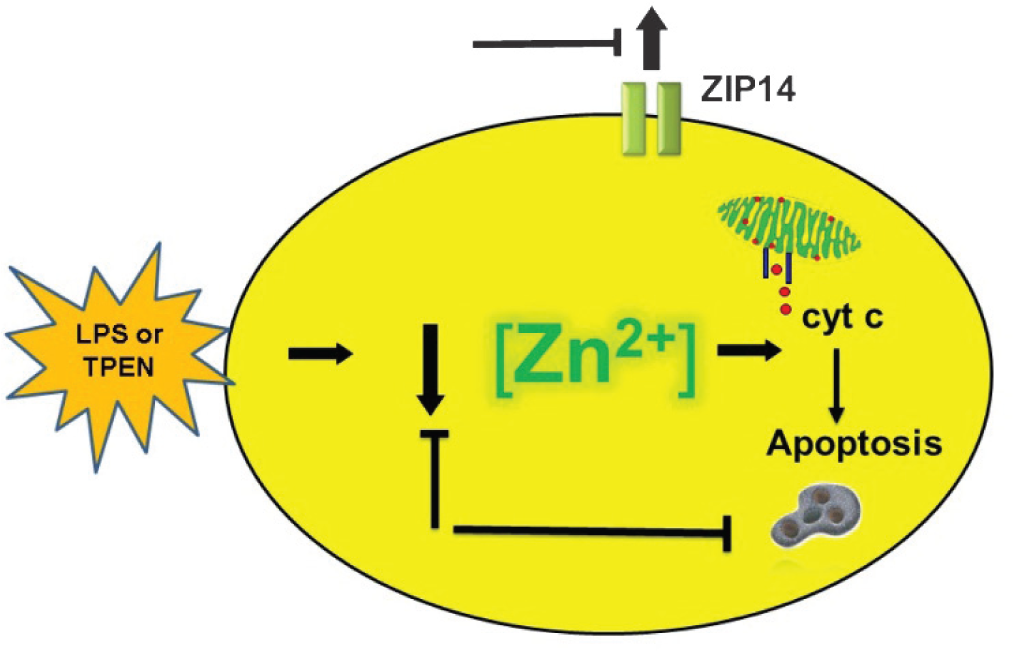
Functional role of labile zinc in LPS-induced apoptosis. LPS caused a decrease in labile zinc in SPAECs (as determined by zinc indicator, FluoZin-3, activity of zinc-sensitive MRE, and changes in steady-state mRNA of zinc importer, ZIP14). The contributory role of decreases in labile zinc in LPS-induced apoptosis (as determined by caspase-3/7 activation, cytochrome c release, and PS externalization) was verified by mimicking the effects of LPS with zinc chelator, TPEN. Blocking LPS- or TPEN- induced decreases in labile zinc inhibited consecutive increase in apoptosis and ZIP14 mRNA providing support for a signaling role of labile zinc in pulmonary endothelium.
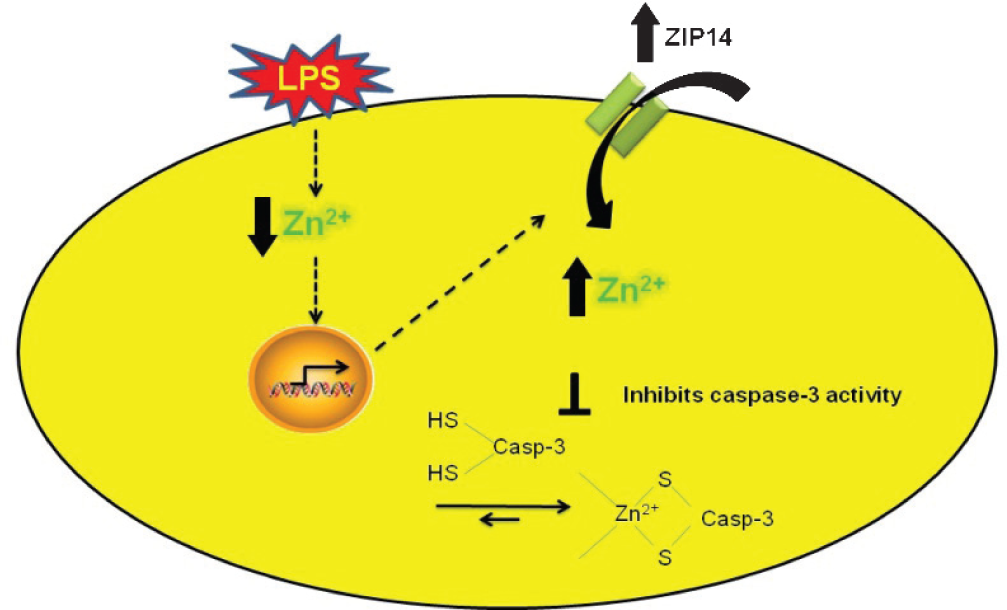
Cytoprotective effect of exogenous zinc is ZIP14 dependent. LPS induced decreases in labile zinc are associated with increases in capsase-3 activity and upregulation of zinc importer, ZIP14 to restore the loss of labile zinc mediated by LPS. Elevation in labile zinc via ZIP14 inhibits apoptosis by inhibiting caspase-3 activity. siRNA to ZIP14 blocked zinc uptake and abrogated zinc mediated protection against LPS-induced apoptosis.
The results we obtained in pulmonary arterial endothelial cells isolated from mature sheep are distinct from those obtained in SPAEC from fetal sheep. We initially noted that addition of large concentrations of zinc to the medium of SPAEC was associated with necrosis.[30] In contrast, elevations in intracellular labile zinc (via addition of exogenous zinc[31] or after exposure to large doses of H2O2[112] or NO[31]) were reported to induce apoptosis in fetal SPAEC. Alternatively, we have consistently noted that chelation of intracellular zinc with TPEN led to dose-dependent apoptosis in mature SPACE whereas a similar maneuver inhibited apoptosis in fetal SPAEC.[31,112]
NO-(MT)-ZN2+ SIGNALING IN PULMONARY ENDOTHELIUM
In aerobic conditions, NO (e.g., presumably via formation of nitrosonium ion intermediate) can S-nitrosate metallothionein[113] and cause the release of Zinquin detectable changes in labile zinc in intact cells.[114] We[49,115] have confirmed these observations and demonstrated the following: (1) S-nitrosation caused conformational changes of MT (via fluorescence resonance energy transfer techniques) in intact pulmonary endothelium consistent with zinc release;[49,116] (2) NO caused an increase in labile zinc in pulmonary artery endothelial cells;[115] and (3) MT was the requisite target for NO resulting in such changes in labile zinc.[115] Subsequent investigations supported the potential for MT to participate in intracellular signal transduction pathways in pulmonary endothelium.
Exposure of mouse lung endothelial cells (MLEC) to the NO donor, S-nitroso-N-acetylpenicillamine (SNAP, 200 μM), caused nuclear translocation of the zinc dependent transcription factor, MTF-1, and such activation was not apparent in MT null cells. Translocation of MTF-1 was associated with NO mediated increase in MT gene expression itself[117] suggested that S-nitrosation of zinc-thiolate clusters in MT and subsequent alterations in zinc homeostasis are participants in intracellular NO signaling pathways affecting gene expression.
We observed that zinc chelation (TPEN) abrogated hypoxic vasoconstriction in isolated perfused mouse lungs (IPL), and that IPL from MT null mice showed significantly less constriction than wild-type controls. Data obtained using NO-sensitive FRET reporters supported both enhanced NO production and S-nitrosation of MT during hypoxic exposure. These events were accompanied by NO-dependent increases in labile zinc (Fluo-Zin-3) in subpleural vessels of MT +/+, but not MT −/− mice. These data supported a role for zinc thiolate signaling in pulmonary vasoregulation. Subsequent studies in cell-based models revealed a link between hypoxia-induced elevations in labile zinc and changes in myosin light chain phosphatase (MLCP) activity, ultimately leading to stress fiber formation and endothelial cell contraction.[118]
Most recently, we showed that zinc chelation abrogates NO-mediated protection against LPS-induced apoptosis.[34] Relative changes in labile zinc after exposure to cytoprotective doses of the NO donor SNAP (250 μM) or exogenous zinc (10 μM) were assessed by Fluozin-3, and a comparable increase in intracellular labile zinc was noted in both conditions.[34] We, further showed that both NO-mediated increases in labile zinc, and NO-mediated protection against LPS-induced apoptosis, are dependent on MT via siRNA to sheep MT isoforms,[34] thus implicating NO-MT-Zn2+ signaling in apoptotic pathways in the pulmonary endothelium (Fig. 4).
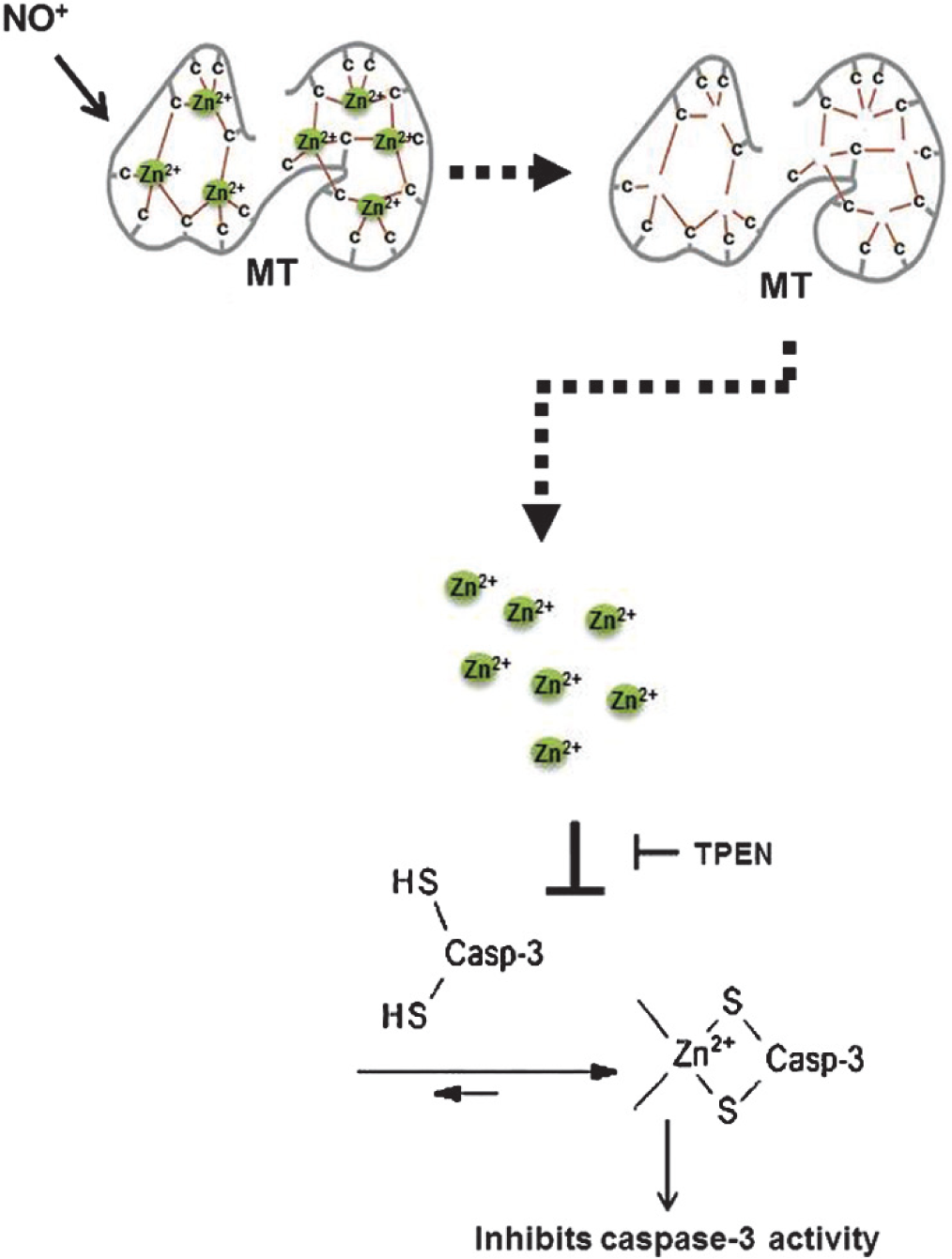
ZINC AS AN EFFECTOR MOLECULE IN PULMONARY ENDOTHELIUM
Decreases in labile zinc have been reported to precede the earliest detectable alterations in cell function,[119] morphology,[119] and apoptosis.[111,119] We and others have reported that chelation of zinc causes spontaneous apoptosis in pulmonary endothelia[30] and epithelia,[4] and elevations in labile zinc via ZIP14[34] or iNOS induced NO or NO donors[111,120–124] inhibits apoptosis in cultured pulmonary endothelial cells. An anti-apoptotic role of zinc has been reported in relation to a variety of stimuli including TNF-α,[125] cadmium,[126] cholesterol,[127] and linoleic acid[33] induced apoptosis. Although the molecular mechanism by which zinc inhibits apoptosis is not clear, several reports suggest that zinc inhibits the following: Ca2+/Mg2+-dependent endonucleases that are responsible for DNA fragmentation;[128] the activity of caspase-3, a critical protease in apoptosis;[129] the processing of caspase-3;[130,131] and bax activation, cytochrome c release, and apoptosome function.[132] Zinc also increases the ratio of Bcl-2 to Bax resulting in the inhibition of caspase activity.[133] We reported the following in SPAECs: decreases in labile zinc mediated by LPS cause casapse-3 activation;[104] LPS-induced caspase-3 activity is sensitive to pan caspase inhibitor;[104] and extracellular zinc inhibits LPS-induced caspase-3 activity.[104,106] We posed a question whether zinc directly binds capsase-3 and modulates its activity. Our results in vitro confirmed that zinc directly inhibits caspase-3 activity.[106] Although NO can S-nitrosate caspase-3 and inhibit its activity,[134] our results suggest that s-nitrosation of MT by NO leads to a release of zinc that is associated with a TPEN dependent cytoprotective caspase-3 inhibition, leading us to suggest that direct S-nitrosation of caspase-3 alone is not likely to account for these results. Collectively, our observation adds to the elegant studies in airway epithelium[12,106] that revealed the following: labile zinc proximity with procaspase-3 prevent the activation of procaspase-3; and zinc depletion activates procaspase-3.[106] These studies provide support for the anti-apoptotic role of labile zinc in the lung.
ZINC HOMEOSTASIS AND ACUTE LUNG INJURY: COMPLEXITIES OF INTEGRATED RESPONSE
Several studies have demonstrated that zinc deficiency sensitizes the lung to acute injury. In particular, dietary restriction led to enhanced sensitivity to polymicrobial sepsis.[36,109] Hyperoxic[135,136] lung injury in mice and macrophage and epithelial cell dysfunction in alcohol fed rats was ascribed to zinc deficiency,[107] Zinc repletion reversed phenotype in all three conditions. Although pulmonary endothelial cell dysfunction may have been a component of all these models, any supportive insight into the cellular contributions of zinc dyshomeostasis to these observations largely relates to background information on zinc in respiratory epithelium.
Nonetheless, hypozincemia in septic or aseptic[59,98] conditions is a somewhat underappreciated phenomenon. Transmigration of zinc from tissues, including lung to liver, has been noted in hyperoxia,[137] bacterial sepsis,[138] and turpentine injury[139] and has been presumed to subserve the following: gluconeogenesis in liver; new protein synthesis in acute phase response; or host defense in an analogous fashion to hypoferremia in bacterial pneumonia.[139] Hepatic expression of ZIP14 appears critical in this phenomenon[140] We recently (in unpublished observations) noted that hepatic expression of metallothionein was important in transmigration of zinc from lung to liver during hyperoxic lung injury apparently contributing to the unexpected observation that MT null mice were resistant to hyperoxia. Collectively, these observations suggest that additional insight into the mechanisms underlying such transmigration may provide new therapeutic targets and strategies and potentially support exogenous zinc as a rational therapeutic agent in acute lung injury.
In summary, compelling evidence is emerging in pulmonary endothelium to complement a larger and growing body of experience in extrapulmonary tissue that labile zinc is a key effector molecule. Critical aspects of the magnitude of labile pool of intracellular zinc accounting for these signaling pathways awaits more refined ratiometric or quantitative fluorescent indicators. Genetic and acquired aspects of zinc dyshomeostasis and deficiencies await further insight into the function and cellular distribution of large family of zinc transporters and metal binding proteins. Nonetheless, it is apparent that the facile and common nature of zinc and nitric oxide chemistry support a role for NO-MT-Zn2+ pathway and the uqibuitous nature of these molecules in sepsis and acute lung injury make them a rational novel therapeutic target.