
Abstract
Myoepithelial neoplasms of the soft tissues are a rare, heterogeneous group of tumors for which classification continues to evolve. While well defined within salivary glands, they can also arise in viscera and soft tissues, where diagnosis is challenging due to the lack of clinical and pathological familiarity. We present the case of a 36 year old man with myoepithelial carcinoma arising as a primary tumor within the soft tissues of the neck, which metastasized to the cecum, causing intussusception. This spindle cell neoplasm showed the classic S100 protein, smooth muscle actin and pancytokeratin-positive immunoprofile. Metastasis of myoepithelial carcinoma to the cecum has not been previously described, and coupled with the spindle cell morphology, may cause significant diagnostic difficulty in the absence of clinical familiarity, particularly as there is morphologic overlap with spindle cell neoplasms arising more commonly in gastrointestinal sites, including gastrointestinal stromal tumor, leiomyosarcoma and sarcomatoid carcinoma.
Keywords
Introduction
Myoepithelial tumors of soft tissue represent a group of neoplasms with morphologic, immunohistochemical and ultrastructural features of myoepithelial differentiation. The marked heterogeneity of these tumors has led to difficulties in their classification, which remains incomplete, although better understanding of their genetics has led to considerable advances in their characterization over the past decade. We describe a case of myoepithelial carcinoma arising in the neck, which metastasized to the cecum three years after resection of the primary neoplasm. Primary and metastatic tumor both showed predominantly spindle cell morphology (a rare but documented histologic pattern) and a classical immunoprofile, with co-expression of S100 protein, smooth muscle actin (SMA) and cytokeratin and loss of nuclear expression of INI1, although the neoplasm did not harbor detectable EWSR1 or FUS rearrangements with fluorescence in situ hybridization (FISH).
Myoepithelial neoplasms share the common feature of differentiation towards myoepithelial cells, but are otherwise a markedly heterogeneous group of tumors displaying prominent morphologic, immunohistochemical and genetic variation. These can arise within organs such as breast and lung, and in skin and subcutis, soft tissue and bone.1–5
Histologically, approximately a third are mixed tumors of either eccrine or apocrine type (morphologically resembling those recognized within salivary glands), while two thirds lack ductular differentiation. 3 Soft tissue myoepithelial tumors occur with a roughly equal gender distribution, and over a wide age range, predominantly in the second to fourth decades,2,5 with about 20% occurring in children.1,2 The most common sites are the extremities and limb girdles, followed by the head, neck and trunk.6,7 There is a spectrum of behavior; histologically benign and low-grade soft tissue myoepithelial tumors have a local recurrence risk of <20%, typically without metastasis, while about 40% of malignant myoepithelial neoplasms recurred and about one third metastasized to lymph nodes, lungs or other sites, 2 including mediastinum, spine, orbit, brain, bone and soft tissues of the thigh. 2 However, metastasis to the cecum or indeed to the bowel has not been previously described.
Histologically, these tend to be lobulated neoplasms with varying growth patterns, including nested, trabecular, fascicular or solid, with cells varying from epithelioid, spindled and clear to plasmacytoid, typically with relatively mild nuclear atypia and mitotic figures rarely in excess of 5 per 10 high power fields. The stroma ranges from collagenous to myxoid or sometimes chondromyxoid, and more rarely there is adipocytic, cartilaginous or bony metaplasia. Histologically malignant features include nuclear pleomorphism with prominent nucleoli, necrosis and atypical mitoses.2,8 Myoepithelial neoplasms have a varied immunoprofile, but generally express S100 protein and pancytokeratins and/or EMA, as well as variable SMA, CD10, calponin, glial fibrillary acidic protein and p63, and occasionally desmin. Loss of nuclear INI1 is seen in about 10% of adult soft tissue myoepithelial carcinomas and 40% of pediatric myoepitheliomas.1,9 Up to 50% of soft tissue myoepithelial neoplasms harbor EWSR1 gene rearrangements (with identified partner genes including POU5F1, PBX1, ZNF444 and ATF1),10–16 and FUS rearrangements are also described. EWSR1-rearranged myoepithelial tumors of deep soft tissues and bone have not shown ductal or glandular differentiation or of cartilage or bony matrix. 10 In contrast to soft tissue myoepithelial tumors with EWSR1 or FUS rearrangements, a proportion of myoepithelial neoplasms of skin and soft tissue with tubuloductal differentiation and mixed tumors of the salivary glands show recurrent PLAG1 rearrangements,17–19 in line with these representing genetically distinct subclasses.
It is likely that myoepithelial tumors have been significantly under recognized previously, due to their varied morphology, histologic and immunohistochemical overlap with a variety of other neoplasms, and the lack of familiarity of physicians with these entities. This case emphasizes the need for awareness of this tumor type, and highlights both an unusually aggressive clinical course and atypical pattern of metastasis to a gastrointestinal site where there is a wide differential diagnosis of neoplasms associated with markedly different management strategies. Recognition of these tumors is also important because of refinements in their genetic characterization, which may lead to targeted therapeutic avenues in future.
Case Report
A 36 year old male had a previous history of primary myoepithelial carcinoma originating from the soft tissues of the right posterior neck (away from salivary glandular parenchyma), which had been treated with radical excision and adjuvant radiotherapy. One year later, he developed bilateral pulmonary metastases, for which he received carboplatin and capecitabine chemotherapy, with which there was progressive disease after two cycles. Therapy was switched to single agent doxorubicin, which resulted in a partial response. Two years later, the patient had a thoracotomy and pulmonary metastatectomy for slowly progressive disease. Three years after diagnosis of the primary tumor, follow up computed tomography (CT) scan showed evidence of ileocolic intussusception, with the lead point presumed to represent a polyp or mucosal metastasis. At diagnostic laparoscopy, the intussusception had resolved, but a mass was evident in the cecal pole, which, with the ascending colon was mobilized laparoscopically as part of a limited right hemicolectomy. Grossly, the specimen comprised a 3×2.8 cm piece of terminal ileum, with 8.5×7.5 cm cecum and attached 12×0.7 cm appendix. On the external surface of the cecum was an area of puckering 5 cm from the distal resection margin. On opening, the area of puckering showed a 5.8×4×2.5 cm solid polypoid brownish-white tumor, which on sectioning was seen to infiltrate the muscularis propria but not the pericecal fat. The appendix, terminal ileum and resection margins were free of tumor.
Histologically, the cecum was infiltrated by a cellular neoplasm which showed similar features to those of the primary neoplasm of the posterior neck soft tissues, and was composed of loose fascicles and sheets of short spindle and epithelioid cells, with mildly pleomorphic ovoid vesicular nuclei and moderate amounts of amphophilic cytoplasm (Figure 1A-D). Occasional larger, rhabdoid-like cells were interspersed. The mitotic index was 8/10 high power fields, and there were prominent areas of necrosis (Figure 1C) (which were also present in the primary neoplasm). The neoplasm ulcerated the cecal mucosa (Figure 1B), extensively infiltrating the muscularis propria (Figure 1D) and extended to pericecal adipose tissue without involving the serosa. No perivascular or perineurial invasion was present. The resection margins, terminal ileum, ileocecal valve, appendix and two lymph nodes within pericecal fat were free of tumor. The tumor showed focal, strong expression of S100 protein, AE1/AE3, epithelial membrane antigen (EMA) (Figure 1E), SMA (Figure 1F) and calponin, while scattered cells were positive for CD34. It was negative for CD117, DOG1, desmin, h-caldesmon and D2-40, with diffuse loss of nuclear expression of INI1 (Figure 1G). CK7, CK20 and CDX2 were also negative, helping to exclude local epithelial origin. The Ki67 proliferation index was high, labeling >70-90% of tumor nuclei (Figure 1H), and this was higher than that seen in the primary tumor, where approximately 40-50% of nuclei had been labeled. FISH using FUS or EWSR1 DNA probes (Vysis, Abbott Laboratories Ltd, Maidenhead, UK) showed no evidence of either EWSR1 or FUS gene rearrangements, and EWSR1-NR4A3, TAF15-NR4A3, EWSR1-FLI1, EWSR1-ERG, EWSR1-WT1, EWSR1-ATF1 and EWSR1-CREB1 fusion transcripts were undetectable by RQ-PCR. The features were consistent with metastatic myoepithelial carcinoma with predominantly spindle cell morphology. The patient is currently well with stable pulmonary disease and without cecal recurrence, five months after resection of the cecal mass.
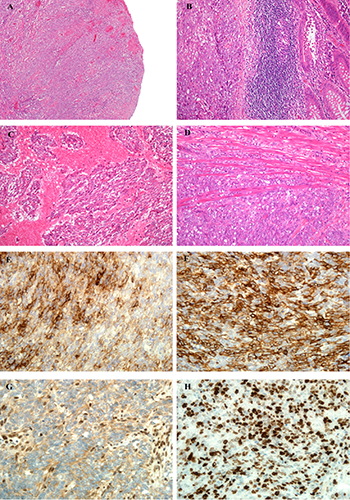
A) Myoepithelial carcinoma. Histologically, the cecum is ulcerated by a prominently nodular neoplasm. The ulcerated surface is covered by fibrin and there is surrounding granulation tissue, but the solid, cellular appearance of the tumor is discernible. B) The tumor is composed of loose fascicles of short spindle and epithelioid cells, with mildly pleomorphic nuclei and moderate amounts of amphophilic cytoplasm. The bowel mucosa (right of field) can be seen to be breached by the neoplasm. C) There are prominent areas of geographic necrosis around viable tumor islands. D) The tumor extensively infiltrates the muscularis propria of the bowel, and is seen to splay the smooth muscle fibers of the bowel wall. E) Immunohistochemistry for epithelial membrane antigen shows focal, strong expression within the neoplasm. F) The tumor also shows strong positivity for smooth muscle actin in most of its cells, supporting a myoepithelial immunophenotype. G) Loss of nuclear INI1 expression is seen in approximately 10% of adult myoepithelial carcinomas, and is a prominent feature of this case. The epithelioid cells of the tumor show diffuse loss of INI1 within nuclei, in contrast to the retention of INI1 expression in the nuclei of normal cells (here, the endothelial cells and lymphocytes act as positive controls). H) The tumor shows a very high Ki-67 proliferation index, and at least 70% of tumor nuclei are labeled in this field.
Discussion
We describe a case of myoepithelial carcinoma metastatic to the cecum in a 36 year old male, three years after excision of a primary neoplasm in the soft tissues of the posterior neck. Myoepithelial neoplasms represent a rare group which is still incompletely characterized clinically, pathologically and genetically, and this case highlights an unusual metastatic pattern of myoepithelial carcinoma and the importance of its clinical diagnostic recognition, particularly as without an appropriate clinical history, these lesions can show significant histologic overlap with a variety of primary spindle cell neoplasms occurring in the gastrointestinal tract which are likely to require significantly different treatment approaches. The cecum is a highly unusual metastatic site for myoepithelial carcinoma, and to our knowledge this location has not been previously documented. The aggressive nature of the neoplasm is also a less typical finding, as myoepithelial tumors occurring in children are more likely to show malignant behavior than those of adults.1,2 Interestingly, myoepithelial neoplasms lacking EWSR1 rearrangements have tended to follow a benign clinical course, 11 although this behavior was not shown by this current case.
The principal differential diagnosis of a spindle cell neoplasm at this site is of gastrointestinal stromal tumor (GIST), sarcomatoid carcinoma, and spindle cell sarcomas including leiomyosarcoma and synovial sarcoma. GIST can display a variety of morphologies, including epithelioid forms but is most frequently composed of fascicles of uniform spindle cells, sometimes with paranuclear vacuolations or palisading. >90% of GISTs express DOG1, CD117 or CD34, usually in a diffuse pattern, whereas myoepithelial neoplasms are essentially negative for these markers. Leiomyosarcoma and myofibrosarcoma are spindle cell neoplasms with smooth muscle and myofibroblastic differentiation respectively, that may occur at this site and can show focal cytokeratin expression, leading to diagnostic confusion with myoepithelial tumors. Leiomyosarcoma typically shows diffuse expression of SMA rather than the more focal expression seen in myoepithelial neoplasms, as well as strong expression of other broad-spectrum myoid or smooth muscle markers desmin and h-caldesmon. Myofibrosarcomas similarly also express SMA, as well as calponin. Some also express desmin, but most lack h-caldesmon. Most synovial sarcomas show focal cytokeratin and EMA expression, up to 30% can express S100 protein and a subset arise intra-abdominally, 19 leading to potential diagnostic confusion with myoepithelial tumors. However, most synovial sarcomas are positive for TLE1 20 as well as bcl-2 and CD99, and they are defined by the presence of a specific chromosomal translocation, t(X;18), leading to the generation of SS18-SSX fusion oncogenes which are absent in all other neoplasms. Sarcomatoid carcinoma can be particularly difficult to differentiate from myoepithelial carcinoma, but patients with sarcomatoid carcinoma will tend to be older and likely to have a history of previous carcinoma. Carcinoma may also show dysplasia or in situ carcinomatous change in the overlying epithelium, or the spindle cell neoplasm may harbor areas of atypical epithelial nests or islands, and immunohistochemistry (e.g. with TTF-1 or CDX2) may indicate potential primary sites. Spindle cell malignant peripheral nerve sheath tumor (MPNST) can also show focal nuclear S100 protein expression, but often occurs in patients with NF1, and may be associated with a nerve or pre-existing benign nerve sheath neoplasm. While its morphology is variable, it typically comprises long fascicles of cells with buckled, tapered or wavy nuclei. Additionally, these neoplasms in the differential diagnosis of myoepithelial tumors will all show nuclear retention of INI1, another helpful distinguishing factor. INI1 is ubiquitously expressed in most cell nuclei, and its loss in nuclei is seen in a relatively limited set of tumors, including a proportion of myoepithelial neoplasms as well as most extrarenal rhabdoid tumors, about 90% of epithelioid sarcomas, half of epithelioid MPNST and some extraskeletal myxoid chondrosarcomas.
The importance of recognition of myoepithelial carcinoma lies in the different clinical management of the tumors in its differential diagnosis. Most patients with metastatic soft tissue sarcoma are treated with palliative systemic therapy. Regardless of histology, patients with solitary or oligometastatic disease may benefit from metastatectomy, to provide symptomatic benefit and potentially prolonged progression free survival. However, the evidence base for metastatectomy consists of retrospective case series with inherent bias. The role of post-operative systemic therapy following complete resection of metastatic disease remains undefined. The selection of systemic therapy for patients with metastatic soft tissue sarcoma is increasingly based on the underlying histologic subtype and molecular characteristics of a given tumor. The introduction of the tyrosine kinase inhibitor, imatinib, has revolutionized the treatment of metastatic GIST. In addition, trabectedin and gemcitabine/docetaxel have shown particular activity in leiomyosarcoma and there are currently trials of MDM2 and CDK4 inhibitors in well-differentiated and dedifferentiated liposarcoma. This is of particular importance in the discussion of our case, as the selection of systemic therapy could be based on the underlying histologic subtype.
Conclusions
In summary, we report a rare case of myoepithelial carcinoma metastasizing to the cecum, from a soft tissue primary of the neck in a 36 year old male. The cecal tumor had the morphologic and immunohistochemical features of its primary, including complete loss of INI1 expression. This highlights an unusual metastatic pattern for this rare neoplasm, and emphasizes the necessity of awareness of this entity and of its morphologic appearances, particularly as the histologic features are heterogeneous, and overlap significantly with tumors more commonly expected at this site, such as GIST, sarcomatoid carcinoma and leiomyosarcoma. Further molecular characterization will contribute to the ongoing classification and prognostication of myoepithelial neoplasms, and most importantly, may shed light on effective therapeutic strategies for aggressive variants.
Footnotes
Acknowledgments
We acknowledge support from the NIHR Royal Marsden/ICR Biomedical Research Centre.