
Abstract
Myoepithelial tumors are extremely rare tumors, with only about 30 cases reported in the literature to date. We present the case of a 27-year-old female diagnosed with malignant right fibula myoepithelial tumor/myoepithelial carcinoma, initially treated with systemic chemotherapy. During treatment, she developed uncontrolled leg pain and metastatic disease to the lungs, leading to a palliative above-knee amputation. Due to the rarity of malignant myoepithelial tumors and the lack of treatment guidelines, a systematic literature review was conducted, exploring various chemotherapeutic and biological agents. Her treatment was planned by a multidisciplinary team, considering her willingness to continue treatment, and ultimately, a decision was made to proceed with palliative care. This case report aims to provide valuable insights for physicians managing patients with this rare diagnosis.
Introduction
The myoepithelial tumor (MET), first described in 1975 by Stromeyer et al., 1 originates from myoepithelial cells primarily located in the salivary glands, skin, and soft tissues. When malignant, it is referred to as malignant MET. 2 Due to its rarity, treatment strategies are case-specific, typically involving surgery, radiation therapy, and chemotherapy. 3 MET has an equal gender distribution, predominantly affecting young to middle-aged adults. 4 Literature reports a recurrence rate of ~40% and a metastasis rate of 22%, with common metastatic sites, including the liver, lungs, soft tissues, and bones.5,6 This case highlights the aggressive nature of MET, the limited treatment options, the diagnostic challenges associated with its identification, the role of immunohistochemistry, and the necessity of a multidisciplinary approach.
Case presentation
A 27-year-old female presented with a 3-month history of right leg pain. Her medical history is unremarkable, with no current medications or family history of malignancy. The complete metabolic panel and complete blood count were normal. Physical examination revealed tenderness, decreased range of motion, and palpable swelling in the right lower leg. MRI showed a 4.5 × 5 × 6.5 cm mass near the fibular head, exhibiting muscle-like intensity on T1-weighted images and increased brightness on Short Tau Inversion Recovery(STIR) images. The mass was found to invade the surrounding muscles and the lateral tibial condyle (Figure 1). A core biopsy revealed epithelioid cells embedded in a myxoid background, with frequent mitosis and necrosis. Muscle invasion was also noted. No osteoid, lipoblasts, multinucleated giant cells, or bony tissue were observed. Immunohistochemistry of the tumor cells showed diffuse positivity for Pan-CK, CK5/6, P63, CK7, EMA (focal), and S100, while being negative for CD99, FLI-1, SATT, vimentin, PAX8, desmin, myogenin, SOX10, chromogranin, GATA3, synaptophysin, SMA, CK20, DOG-1, GCDFP, CD34, and INI1/BAF47 (Figure 2). Genetic and molecular testing confirmed proficient MMR status, and wild-type BRAF with negative results for PD-L1, ER, PR, Her2, ALK, and ROS1. These findings support the diagnosis of malignant MET.

MRI revealed an abnormal mass measuring 4.5 × 5 × 6.5 cm centered around the fibular head in the right proximal fibula.

(a) Myoepithelial carcinoma ×4; (b) myoepithelial carcinoma ×10. (c–f) Microphotograph showing immunohistochemical results: Pan-CK protein positivity, P63 positivity, S100 positivity, and actin (SMA) focal positivity, respectively.
Computed Tomography (CT) scans of the brain, neck, chest, and abdomen were unremarkable. A PET–CT scan revealed a heterogeneous intensely hypermetabolic malignant mass with FDG uptake up to Standardized Uptake Value (SUV) max 21.6, the mass likely originating from the right proximal fibula measuring 6 × 6 × 5.5 cm with cortical destruction and muscular invasion (Figure 3). A bone marrow biopsy showed no signs of malignancy.
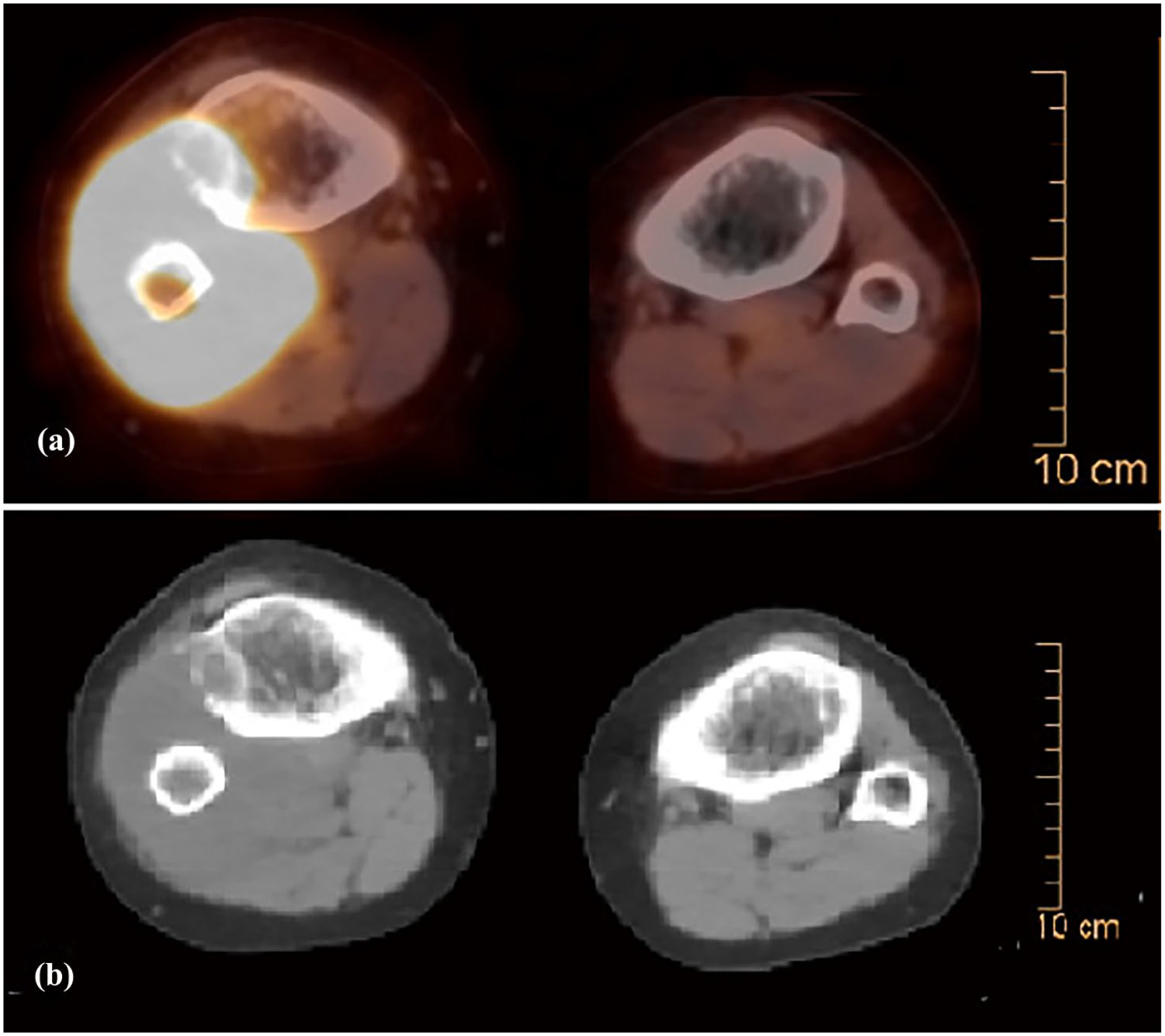
(a) A PET–CT scan revealed a heterogeneous intensely hypermetabolic malignant mass with FDG uptake up to SUV max 21.6, the mass likely originating from the right proximal fibula, measuring 6 × 6 × 5.5 cm with cortical destruction and muscular invasion. (b) The CT part of the PET scan.
The patient’s chemotherapy regimen initially consisted of ifosfamide and etoposide. However, due to the development of neutropenic fever and tumor growth, the treatment was switched to five cycles of vincristine, doxorubicin (Adriamycin), and cyclophosphamide. A reevaluation CT scan revealed bilateral metastatic lung nodules (Figure 4). Subsequent imaging revealed new pulmonary nodules and lymphatic involvement, indicative of disease progression. A PET scan follow up done and showed marked morphological progression with decrease metabolic activity regarding the heterogeneous intensely hypermetabolic right proximal fibula mass, destroying both tibia and fibula associated with muscle atrophy, the mass measuring about 17 cm in longest axial dimension with SUV mas up to 13 (Figure 5). She underwent an above-knee amputation and continued chemotherapy with carboplatin and paclitaxel, followed by irinotecan and temozolomide, and subsequently, gemcitabine and bevacizumab. Despite these treatments, further disease progression was observed. Ultimately, following discussions with her family, she transitioned to palliative care.

A CT scan revealed bilateral metastatic lung nodules.

(a) A PET scan follow-up done and showed marked morphological progression with decreased metabolic activity regarding the heterogeneous intensely hypermetabolic right proximal fibula mass, destroying both tibia and fibula, associated with muscle atrophy, the mass measuring about 17 cm in longest axial dimension with SUV mass up to 13. (b) The CT part of the PET scan.
Discussion
This case presents a rare and aggressive primary MET of the proximal fibula in a young female. Despite early intervention with chemotherapy and surgery, the tumor rapidly progressed, metastasized to the lungs, and ultimately necessitated limb amputation. The patient’s resistance to multiple treatment regimens and the absence of actionable molecular targets highlight the clinical challenges associated with osseous MET and underscore the need for further research into effective therapeutic strategies.
METs are uncommon neoplasms that can arise in both soft tissue and bone and are classified as benign (myoepitheliomas) or malignant (carcinomas). 7 Primary osseous METs were formally recognized in 2002 8 and remain extremely rare, with only 30 cases reported by 2017. 7 These tumors affect individuals across a wide age range and both sexes. Although children may have a higher risk of malignant behavior, adults tend to show lower recurrence rates following surgical resection. 4
In our case, imaging findings were consistent with reported MET characteristics—lytic bone lesions with defined borders and heterogeneous enhancement on MRI.7,9 However, diagnosis remains challenging due to histological variability, necessitating immunohistochemistry for confirmation. 10 Malignant METs are typically marked by infiltrative growth, necrosis, mitotic activity, and nuclear atypia.10,11 Our patient’s tumor expressed Pan-CK, CK5/6, CK7, P63, and S100, with focal EMA positivity (Figure 2), supporting the diagnosis. 4 Rather than listing marker incidence rates, it is more clinically relevant to emphasize that dual expression of epithelial (e.g. EMA or AE1/AE3) and myoepithelial markers (e.g. S100 or GFAP) is essential for diagnosis. 12 Moreover, nuclear atypia and prominent nucleoli indicate malignant potential and a worse prognosis.3,7
What makes this case particularly unique is the tumor’s aggressive biology despite the absence of common therapeutic targets. Molecular profiling revealed Mismatch Repair (MMR) proficiency and negative expression of PD-L1, ALK, BRAF, and other potential markers. These features likely contributed to the limited response to conventional and systemic therapies. Although immunotherapy and targeted therapies have shown efficacy in select soft tissue sarcomas, their role in MET is poorly defined, especially in the absence of immunogenic markers. 10
The rapid progression in this patient raises the possibility of underlying molecular drivers not yet fully understood. Factors such as a high proliferative index, the presence of necrosis, and muscle invasion could indicate an inherently aggressive phenotype. Comparatively, previously reported cases in young adults often exhibit slower progression and better response to treatment, suggesting heterogeneity in tumor biology. 4 A summary of published case reports on myoepithelioma of long bones is presented in Table 1.
Summary of previously published case reports on myoepithelioma of long bone.

NA: not available; F: female; M: male; GFAP: glial fibrillary acid protein.
This case emphasizes the limitations of current therapeutic options for osseous MET and the urgent need for expanded molecular profiling, case registries, and multicenter studies. Early integration of palliative care should also be considered in aggressive, treatment-refractory cases.
Conclusion
This case presents a uniquely aggressive MET originating in the proximal fibula of a young adult, demonstrating rapid progression, early metastasis, and resistance to multiple chemotherapy regimens. The rarity of primary osseous MET, combined with the absence of actionable molecular targets, highlights the diagnostic and therapeutic challenges clinicians face. This report emphasizes the importance of early molecular profiling, multidisciplinary care, and consideration of emerging treatments such as immunotherapy and targeted therapies, even in the absence of established protocols. Future research should focus on identifying prognostic biomarkers and developing standardized treatment strategies to improve outcomes in this rare and aggressive tumor type.
Footnotes
Acknowledgements
The authors would like to express their gratitude to the doctors in the radiology and pathology departments at Augusta Victoria Hospital for their invaluable assistance in collecting patient data.
Ethical considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent for publication
Written informed consent was obtained from the patient(s) for the publication of their anonymized information in this article.
Author contributions
Tasneem AbuShaikha and Saja Alzboun: conceptualization, methodology, writing—original draft. Yazan Haddar, Ameer Alhosheyeh, and Imad Aljaafreh: data curation, writing—review and editing. Ahmad Alhalabia, Haytham Shaban, and Ahmed Barbarawi: resources, validation. Farah Awad and Elias Edward Lahham: supervision, data curation, final approval of manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data used to support the findings of this study are included within the article.