
Abstract
Primary thyroid lymphoma is a rare malignancy, representing 2–8% of all thyroid malignancies and 1–2% of all extranodal lymphomas. The majority of cases concern non-Hodgkin's lymphoma of B cell origin, following by Hodgkin's disease, T cell lymphomas and rarely marginal zone B-cell mucosa-associated lymphoid tissue (MALT) lymphomas. MALT lymphomas have been associated with long-standing autoimmune Hashimoto's thyroiditis. We present the case of a 44-years-old woman with thyroid MALT lymphoma in the background of multinodular goiter of autoimmune origin.
Introduction
Primary thyroid lymphoma represents 2–8% of all thyroid malignancies and 1–2% of all extranodal lymphomas.1–3 Most thyroid lymphomas are non-Hodgkin's lymphomas of B cell origin, following by Hodgkin's disease, T cell lymphomas and rarely marginal zone B-cell mucosa-associated lymphoid tissue (MALT) lymphomas.1–3 MALT lymphomas, which account for 25% of primary lymphomas, arise mainly in the stomach (60–70%), but they also can be found in organs such as the orbit, salivary gland, lung, intestine, liver and thyroid gland. 2 Normally, thyroid gland is devoid of lymphoid tissue while this is reversed during the course of autoimmune diseases such as Hashimoto's thyroiditis.1–3 It has been evident that thyroid MALT lymphomas occur in 0.5% of the cases with Hashimoto's thyroiditis, even after a long period of lymphocytic infiltration which reaches as long as 30 years. A concurrence with papillary thyroid carcinoma has been also reported4,5 The clinical presentation include an enlarging neck mass, dysphagia, hoarseness, dyspnea and usually the patients are euthyroid. Most often the diagnosis is difficult, based either on the morphological or clinical characteristics and flow cytometry, Southern blotting or PCR need to be performed.6–8
Herein, we describe the case of a 44-years-old female who was diagnosed with thyroid MALT lymphoma in the background of autoimmune thyroiditis.
Case Report
A-44 years-old, obese, woman, with autoimmune thyroiditis on L-thyroxine treatment (100 ug/d), was referred to our department for further evaluation of a rapidly growing mass of the right lobe of the thyroid gland, associated with gradually increased pain and dyspnea. The laboratory evaluation of the patient revealed normal TSH levels and positive thyroid antibodies, indicating a properly treated autoimmune thyroiditis. A Doppler ultrasound of the thyroid gland revealed multiple hypoechoic nodules (max. diam 2 cm) with microcalcifications and increased peripheral and central vascularity. A fine needle aspiration was performed, which showed suspicious for malignancy findings of the nodule while a lymphoproliferative infiltration of the thyroid parenchyma and histiocytes, mainly with epithelioid morphology, were also observed. Her sister and her mother had the diagnosis of occult papillary thyroid cancer on the background of autoimmune thyroiditis, which had been diagnosed at the age of 35 years.
Due to the suspicious ultrasound and cytologic findings, the positive for thyroid cancer family history and the difficulty of having a systematic follow up, total thyroidectomy was performed. The pathology showed thyroid MALT lymphoma on the background of autoimmune Hashimoto's thyroiditis. More specifically, the H&E staining, in addition to chronic lymphocytic thyroiditis, showed a characteristic infiltration of the thyroid tissue with monoclonal plasma cells and big epithelial histiocytes. The immunohistochemical study revealed monoclonal cytoplasmic immunoglobulin expression of cIg-λ in the plasma cell population (Figure 1).
A) Hematoxylin and eosin (H&E) staining showing mucosa-associated lymphoid tissue (MALT) thyroid lymphoma; B) immunohistochemical evaluation reported MALT-balls; C) D) E) F) G) serial sections immunostained for CD79+, CD20+, cIg-κ, cIg-λ, L26, respectively.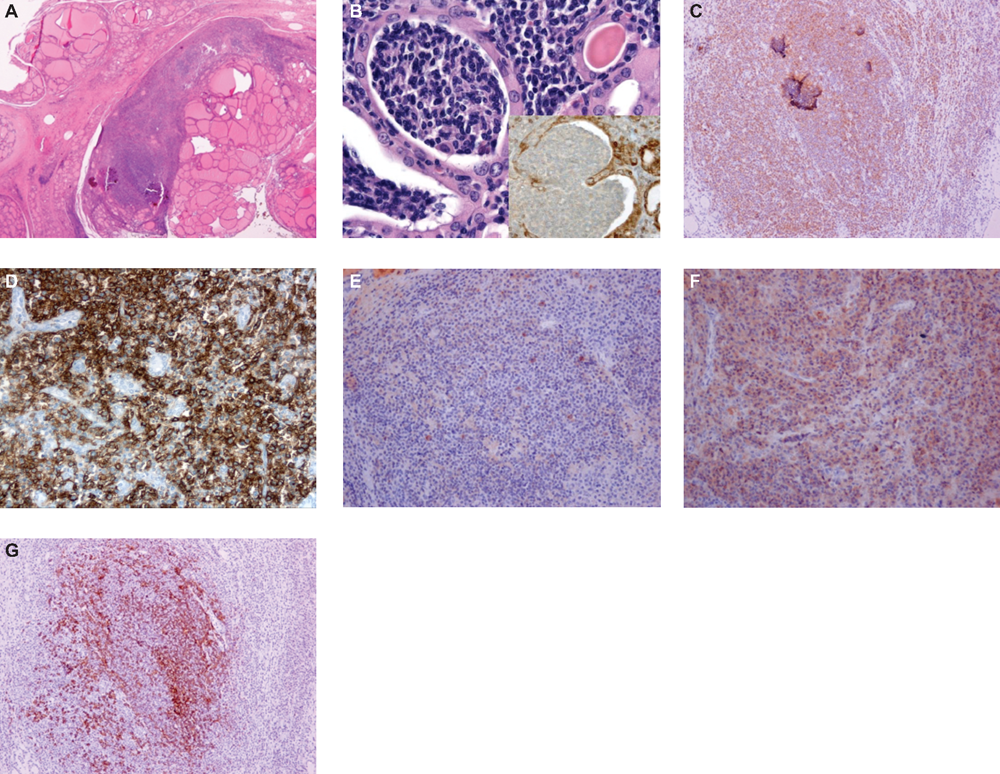
The patient underwent further evaluation with gastroscopy and scanning tomography of the neck, thorax and pelvis which were all negative. Bone marrow biopsy reported a reactive marrow and the immunohistochemical study (anti-CD3/CD20/CD138, Glycoporin A, κ and λ light chain immunoglobulin) was negative for the presence of MALT lymphoma.
The potential benefits of being further treated by radiotherapy and/or chemotherapy, were discussed with the oncologists and the final decision was the annual follow up. Two years after she is well, with no evidence of local or systematic disease, adequately replaced with L-thyroxine
Discussion
The present report, refers to a patient with thyroid MALT lymphoma on the background of autoimmune thyroiditis.
Primary thyroid MALT lymphoma is a quite rare neoplasm accounting for 2–8% of all thyroid alignancies and 1–2% of all extranodal lymphomas.1–3 It tends to present in women during their seventh decade of life, while our case refers to a middle-aged woman. Men with thyroid MALT lymphoma, exhibit a worse prognosis, compared to the well differentiated thyroid cancer.1–3,9
Most commonly, it presents as a painless rapidly growing thyroid mass, as in our case, while general symptoms like fever, nocturnal sweating and weight loss mostly suggest secondary thyroid infiltration by lymphoma of another organ.1–3,8,9
Although H. pylori infection has been associated with gastric MALT lymphoma in addition to coronary artery disease and type 1 and 2 diabetes mellitus10–13 this relationship has not been adequately addressed for thyroid MALT lymphoma. The existing data support that autoimmune thyroiditis seems to be a risk factor for the development of thyroid MALT lymphoma due to an acquired pathological transformation of the intrathyroidal lymphoid tissue. 14 Most often the concurrence of thyroid MALT lymphoma with autoimmune thyroiditis creates a lot of problems in its diagnosis. In autoimmune thyroiditis the two main microscopical findings are lymphocytic infiltration of the stroma and oxyphilic change of the follicular epithelium. On the other side, the characteristics of thyroid MALT lymphoma include the atypical lymphoid cells originated within the marginal zone of the lymphoid follicles and extended into the interfollicular space, the thyroid follicular epithelium and the germinal centers. In certain cases, immunocytochemistry (CD 20, CD 43, λ- and κ- light chains), but also flow cytometry and PCR, are needed to confirm the diagnosis of thyroid MALT lymphoma.7–9,15
A lot of questions have been raised regarding the follow up of patients with thyroid MALT lymphomas. The first step consists of a careful systematic evaluation of the patient in order to characterize the thyroid MALT lymphoma as primary. This includes a thorough physical examination, a full biochemical investigation, evaluation of the gastrointestinal tract (gastroscopy, colonoscopy), a bone marrow biopsy and scanning of the neck, chest and abdomen. The second step is the staging of the tumor according to the system proposed by Ann Arbor and modified by Myssoff.7,16,17
Many dilemmas have been raised regarding the proper management of patients with thyroid MALT lymphoma. Retrospective reports suggest an indolent behavior and excellent clinical prognosis for this subset of thyroid lymphomas, leading to the conclusion that single modality therapy with either surgery or radiation may have a treatment role. However, in the absence of randomized clinical trials to compare different treatment options (thyroidectomy, radiotherapy, chemotherapy) in patients with thyroid MALT lymphoma, there are not widely accepted guidelines and thus therapeutical protocols should be planned in an individual base. Total thyroidectomy is the treatment of choice in patients with compressive goiter and localized thyroid MALT lymphoma. Radiotherapy is used as primary therapy in patients with stage IE and IIE and as adjuvant therapy for those with suspected residual disease after thyroidectomy. Chemotherapy is usually indicated for patients with disease stages IIIE and IV but prognostic data are unavailable because of the rarity of this condition. In patients with localized disease, as in our patient, the preferred treatment is total thyroidectomy while the benefit from applying radiotherapy in a total dose of 40 Gy, or chemotherapy, is questionable and with its toxicity being a major issue.1,2,6,8,17–20
In conclusion, our case of a concurrence of primary thyroid MALT lymphoma and autoimmune thyroiditis, is trying to emphasize the need of a careful evaluation of any palpable thyroid mass. This could be a benign lesion but also a malignant lesion, including not only neoplasms of thyroid tissue but also metastatic from other sites or primary neoplasms of extrathyroidal origin, as the case of lymphomas and rarely MALT lymphomas. Moreover, this case in addition to the existing published cases, illustrates that the nature of follow-up care and long-term results of treatment of patients with primary thyroid MALT lymphomas are not fully established.