
Abstract
Abdominal aortic aneurysm (AAA) is a potentially lethal disease associated with immune activation-induced aortic degradation. We hypothesized that xenotransplantation of human adipose-derived stem cells (hADSCs) would reduce aortic inflammation and attenuate expansion in a murine AAA model. Modulatory effects of ADSCs on immune cell subtypes associated with AAA progression were investigated using human peripheral blood mononuclear cells (hPBMNCs) cocultured with ADSCs. Murine AAA was induced through elastase application to the abdominal aorta in C57BL/6 mice. ADSCs were administered intravenously, and aortic changes were determined by ultrasonography and videomicrometry. Circulating monocytes, aortic neutrophils, CD28− T cells, FoxP3+ regulatory T cells (Tregs), and CD206+ M2 macrophages were assessed at multiple terminal time points. In vitro, ADSCs induced M2 macrophage and Treg phenotypes while inhibiting neutrophil transmigration and lymphocyte activation without cellular contact. Intravenous ADSC delivery reduced aneurysmal expansion starting from day 4 [from baseline: 54.8% (saline) vs. 16.9% (ADSCs), n = 10 at baseline, n = 4 at day 4, p < 0.001], and the therapeutic effect persists through day 14 (from baseline: 64.1% saline vs. 24.6% ADSCs, n = 4, p < 0.01). ADSC administration increased aortic Tregs by 20-fold (n = 5, p < 0.01), while decreasing CD4+CD28− (-28%), CD8+CD28− T cells (-61%), and Ly6G/C+ neutrophils (-43%, n = 5, p < 0.05). Circulating CD115+CXCR1−LY6C+-activated monocytes decreased in the ADSC-treated group by day 7 (-60%, n = 10, p < 0.05), paralleled by an increase in aortic CD206+ M2 macrophages by 2.4-fold (n = 5, p < 0.05). Intravenously injected ADSCs transiently engrafted in the lung on day 1 without aortic engraftment at any time point. In conclusion, ADSCs exhibit pleiotropic immunomodulatory effects in vitro as well as in vivo during the development of AAA. The temporal evolution of these effects systemically as well as in aortic tissue suggests that ADSCs induce a sequence of anti-inflammatory cellular events mediated by paracrine factors, which leads to amelioration of AAA progression.
Keywords
Introduction
Abdominal aortic aneurysm (AAA) is a multistep, initially asymptomatic process of excessive aortic inflammation-induced deterioration leading to progressive aortic dilation, aortic rupture, and patient mortality1–3. The risk of aneurysm rupture increases exponentially with the aortic diameter and is associated with mortality of more than 80% despite emergency surgical repair 4 . Unfortunately, no therapeutic options have been demonstrated to retard the growth of aneurysms, so current treatment is limited to elective or emergent endovascular or open surgical repair of larger aneurysms5–8. Excessive aortic inflammation is a key pathogenic factor responsible for the development of aneurysms9–11. It is characterized by activation of circulating monocytes12,13; release of proinflammatory cytokines14,15; local aortic infiltration by neutrophils, macrophages, and lymphocytes16–18; and subsequent degradation of the extracellular matrix (ECM) by macrophage- and neutrophil-derived proteases19–23.
Among the several immune cells involved in the pathogenesis of AAA, subtypes of T cells, in particular, have been shown to play key roles in early aortic inflammation, which promotes degradation of the ECM in AAA24–26. Specifically, AAA patients with small aneurysms, early in their clinical course, have a high prevalence of circulating as well as aortic CD4+CD28− and CD8+CD28− T cells 27 and a paucity of anti-inflammatory CD4+CD25+Foxp3+ regulatory T cells (Tregs)28. Similarly, in animal models, Treg depletion or deficiency has been shown to exacerbate aneurysm formation, whereas viable Treg supplementation inhibits AAA development 29 . Overall, these observations highlight the importance of adequate Treg activity to balance CD28− T cells in AAA initiation and development.
In the past decade, administration of mesenchymal stem cells (MSCs) has emerged as a promising therapeutic modality to modulate inflammatory responses in a wide range of pathologies. Bone marrow (BM)-and placenta-derived MSCs have shown promise in limiting overall AAA expansion in animal models, although the precise modulation of monocytes, M2 macrophages, Tregs, and CD28− T cells by MSCs has not been thoroughly investigated30–35. More recently, adipose-derived MSCs or adipose-derived stem cells (ADSCs) have been recognized as readily available in adequate numbers to permit practical autologous treatment. ADSCs derived from liposuction can be immediately administered upon isolation at the point of care or after only a few days of culture. ADSCs have been reported to induce Treg phenotype and suppress lymphocyte proliferation in vitro36–38. However, their paracrine modulation of transendothelial neutrophil migration or immune subtypes has not been explored, particularly in the context of AAA.
In this article, we determined that ADSCs modulate neutrophil transmigration as well as lymphocyte, monocyte, and macrophage phenotypes both in vitro and in vivo. Moreover, ADSC administration limited topical elastase-induced aortic inflammation and aneurysm expansion in our murine model. We attributed these effects to paracrine factors released from ADSCs transiently trapped in the lung without aortic engraftment.
Materials and Methods
All procedures for collecting human tissues (peripheral blood and adipose tissue) were approved by the Indiana University School of Medicine Institutional Review Board (IRB).
Human Adipose-Derived Stem Cells
Human ADSCs (hADSCs) were isolated by the subcutaneous liposuction of adipose tissue, as previously described 39 . Briefly, adipose tissue was digested in collagenase type I solution (Worthington Biochemical, Lakewood, NJ, USA) with agitation for 1 h at 37°C followed by 250 μm Nitex filtration (Sefar America, Buffalo, NY, USA) and centrifugation at 300 × g for 8 min to separate the stromal cell fraction (pellet) from adipocytes. The stromal fraction was then suspended in cell lysis buffer [154 mM NH4Cl, 10 mM KHCO3, and 0.1 mM ethylenediamine-tetraacetic acid (EDTA); Thermo Fisher Scientific, Waltham, MA, USA] for 5 min at 37°C and centrifuged at 300 × g for 5 min. The stromal pellet was suspended and cultured in microvascular endothelial cell growth medium-2 (EGM-2MV) media (Lonza, Allendale, NJ, USA) and passaged at 60%–80% confluence. ADSCs with Hoechst labeling were used (Invitrogen, Carlsbad, CA, USA) at passages 3 to 5. The hADSCs were suspended in phosphate-buffered saline (PBS; Thermo Fisher Scientific) for use in vivo. ADSCs isolated using this method were positive for CD90, CD73, CD105, and CD44 and negative for CD106, CD45, and CD31, as described previously 40 .
Human ADSC conditioned media (ADSC-CM) was generated by 20 ng/ml recombinant human tumor necrosis factor-α (TNF-α; R&D Systems, Minneapolis, MN, USA) activation for 24 h. Following this activation, cells were washed and allowed to recover for an additional 24 h in fresh endothelial growth basal medium (EBM2) (Lonza) before ADSC-CM collection.
In Vitro Experiments
To isolate human neutrophils and peripheral blood mononuclear cells (PBMNCs), peripheral blood was isolated from healthy donors and processed with Ficoll-Paque Plus (GE Healthcare Life Sciences, Pittsburgh, PA, USA), according to the manufacturer's instructions. Briefly, blood plasma was removed, and the leukocyte-rich upper layer was transferred and diluted with 30 ml of Hank's balanced salt solution (HBSS; Thermo Fisher Scientific). The suspension was then layered onto 15 ml of Ficoll-Paque Plus (GE Healthcare Life Sciences) and centrifuged into its respective layers: plasma, PBMNCs, Ficoll-Paque Plus, neutrophils, and erythrocytes. The PBMNC and neutrophil layers were collected and incubated in erythrocyte lysis buffer for 10 min, washed twice, and immediately used.
To study macrophage polarization, 1 × 106 hPBMNCs were incubated with 3 × 104 or 3 × 105 ADSCs on Transwell inserts for 3 days with or without 20 ng/ml TNF-α. Cells that adhered to the bottom of the plate were then isolated and resuspended for fluorescein isothiocyanate (FITC)-CD68 and phycoerythrin (PE)-CD206 (BD Biosciences, San Jose, CA, USA) flow cytometry, as previously described 41 .
To study Treg induction, hPBMNCs were incubated with ADSCs on Transwell inserts for 5 days. Cells that adhered to the bottom plate were then isolated and resuspended for FITC-CD4, allophycocyanin (APC)-CD25, and PE-FoxP3 (BD Biosciences) flow cytometry.
Neutrophil transmigration was determined using Transwell plating in which inserts coated with a confluent monolayer of human umbilical cord endothelial cells (HUVECs) were placed above a bottom chamber with human ADSCs. CellTraceTM carboxyfluorescein succinimidyl ester (CFSE; Invitrogen)-labeled human neutrophils were loaded into the upper chamber, and migration was monitored in response to 4–6 h of human TNF-α (20 ng/ml) treatment (Eclipse Ti; Nikon, Melville, NY, USA).
To measure lymphocyte proliferation, hPBMNCs were stimulated for 6 days at 37°C/5% CO2 with mouse antihuman CD3 (1 μg/ml; BD Biosciences) and mouse antihuman CD28 (0.5 μg/ml; BD Biosciences). [3H]Thymidine (0–5 mCi/well) was then added 18 h prior to measuring [3H]thymidine incorporation.
Animals
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Indiana University. To generate our AAA model, 8- to 12-week-old male C57BL/6 mice (Harlan, Indianapolis, IN, USA) were anesthetized with isoflurane (Thermo Fisher Scientific), and a midline abdominal incision was made to expose the abdominal aorta between the renal artery and the iliac bifurcation. An aneurysm was induced by placing an absorbable gelatin sponge (Pfizer, New York City, NY, USA) containing 47 U/ml porcine elastase solution (Sigma-Aldrich, St. Louis, MO, USA) to the top of the exposed aorta (Fig. 1). The abdominal wall and skin were then closed with sutures. Two hours after surgery, mice were administered either vehicle (PBS) or 106 Hoechst-labeled hADSCs through tail vein injection42,43. Changes in the abdominal aorta were determined by ultrasound (VisualSonics, Ontario, Canada) at baseline and on days 1, 4, 7, and 14. The diameters were objectively measured by the same examiner blinded to the treatment assignment.

Time-dependent topical elastase-induced murine aneurysm formation. (A) Representative videomicrometry of progressive abdominal aortic expansion following topical elastase treatment. (B) A linear correlation between ultrasound- and videomicrometry-measured aortic diameter (n = 22, R2 = 0.8473).
Histology and Immunohistochemistry
Animals were systemically perfusion fixed through the left ventricle using 4% paraformaldehyde (PFA; Sigma-Aldrich). The harvested abdominal aorta, between the renal artery and the iliac bifurcation, was further fixed overnight in 4% PFA followed by paraffin embedding for sequential sectioning (Fig. 2). The sequential sections were stained with either hematoxylin and eosin (H&E; Thermo Fisher Scientific), Masson's trichrome (Sigma-Aldrich), or Verhoeff–VanGieson (Sigma-Aldrich) for inflammatory cell infiltration, collagen, and elastin content, respectively.

Hematoxylin and eosin (H&E) staining of the abdominal aorta was performed on sequential sections. Abdominal aorta between the renal artery and iliac bifurcation was harvested, fixed, paraffin embedded, and sequentially sectioned with 300 μm distance between sections. H&E staining demonstrated a sinusoidal pattern of aortic wall thickening and inflammation reflecting their proximity to the aneurysmal center.
Aortic M2 macrophage and Treg levels were determined by immunohistochemistry as previously described44,45. Briefly, aortic sections were deparaffinized and ethanol rehydrated followed by citrate buffer (Thermo Fisher Scientific)-mediated antigen retrieval. Once processed, the sections were blocked with A protein block (Abcam, Cambridge, MA, USA) and exposed to either: 1:100 rabbit polyclonal CD68 (Abcam), 1:200 rabbit polyclonal CD206 (Biorbyt, Berkeley, CA, USA), 1:200 rabbit polyclonal CD3 (Abcam), or 1:200 rat anti-mouse FoxP3 (eBiosciences, San Diego, CA, USA). Sections were developed using the biotin–avidin/streptavidin system (Vector Laboratories, Burlingame, CA, USA). Cells were counterstained with peroxidase substrate 3,3′-diaminobenzidine (DAB; Sigma-Aldrich) and/or hematoxylin (Sigma-Aldrich).
RNA Extraction and Quantitative Real-Time PCR
Warm saline was flushed through the left ventricle of the animals, after which the abdominal aorta was rapidly excised and snap frozen in liquid nitrogen. Total RNA was extracted from aortic tissue homogenates using the RNeasy mini kit (Qiagen, Valencia, CA, USA) following the manufacturer's instructions. Reverse transcription was performed with the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) and real-time polymerase chain reaction (PCR) with the SYBR Green Master Mix (Applied Biosystems). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control 46 .
Flow Cytometry
Freshly isolated mouse aortic tissue was minced and incubated for 15 min with 37°C collagenase type IA (1 mg/ml; Sigma-Aldrich) dissolved in PBS with 0.5% bovine serum albumin (BSA; GE Healthcare) and 2 mmol/L EDTA. Cell viability was determined by trypan blue (Thermo Fisher Scientific) light microscopy followed by anti-mouse CD16/CD32 (1 μg/ml; eBiosciences) blocking and antigen labeling for CD115, CD25, FoxP3 (eBiosciences), Ly6C, CD4, CD8, CD28, Ly6-G/C (BD Biosciences), or CX3CR1 (R&D Systems). Flow cytometry was performed on a Guava 8HT (Millipore, Billerica, MA, USA). Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Human Alu Sequence DNA Extraction and Real-time PCR
The right lungs of mice treated with hADSCs were harvested at day 1 or 4. Genomic DNA was isolated using the Wizard Genomic DNA purification kit (Promega, Madison, WI, USA) following the manufacturer's instructions. Human Alu sequence real-time PCR was performed as previously described 43 . A standard curve for hADSCs was generated by adding serial ADSC dilutions to control mouse lung tissue before homogenization and DNA extraction.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software (La Jolla, CA, USA). Data are presented as mean ± standard error of the mean (SEM). Differences between groups were examined using Student's unpaired t-tests or two-way analysis of variance (ANOVA). Post hoc Newman–Keuls tests identified differences between groups. Values of p < 0.05 were considered significant.
Results
Human ADSCs Induce In Vitro M2 Macrophage Polarization and Treg Phenotype, Inhibit Neutrophil Transmigration, and Suppress Lymphocyte Activation Through Paracrine Mechanisms
Excessive inflammation is one of the defining characteristics of AAA. It is associated with prominent M1 macrophage polarization, Treg depletion, neutrophil transmigration, and lymphocyte activation 2 . We found that, indeed, hADSCs were able to modulate these inflammatory cellular events in vitro in a dose-dependent manner. Human PBMNC-derived macrophages were cocultured in Transwells with hADSCs for 3 days. Following incubation, the addition of TNF-α induced a significant 83% reduction in CD206+ M2 macrophages when compared to nontreated controls. Importantly, this was reversed in a dose-dependent manner by ADSC coculture (-58% vs. control in 3 × 104 ADSC group, and +116% vs. control in 3 × 105 ADSC group) (Fig. 3A and B). In parallel experiments, the presence of ADSCs induced a 2.5-fold increase in anti-inflammatory Tregs by day 7 (Fig. 3C and D). Additionally, we employed TNF-α as a chemoattractant to induce neutrophil transmigration across a confluent Transwell monolayer of HUVECs. Results demonstrated that ADSCs placed in the bottom of a Transwell reduced neutrophil transmigration by nearly 70% (Fig. 3E–G).

Human ADSCs induce M2 macrophages, inhibit neutrophil transmigration, and suppress lymphocyte activation in vitro. (A) Adipose-derived stem cells (ADSCs) increased the percentage of M2 macrophages (CD14+CD206+/total CD14+) in human peripheral blood-derived mononuclear cell (PBMNC) 3-day Transwell cultures with or without tumor necrosis factor-α (TNF-α) in a concentration-dependent manner. (B) Representative flow cytometry plot. (C) ADSCs cocultured with human PBMNCs had an increased percentage of regulatory T cells (Tregs). (D) Representative CD4-gated flow cytometry plot. (E) Neutrophil transmigration assay cartoon. (F) ADSCs inhibited TNF-α-induced neutrophil migration across an endothelial cell (EC) monolayer. (G) Carboxyfluorescein succinimidyl ester (CFSE)-labeled neutrophils. (H) ADSCs and ADSC-conditioned media (ADSC-CM) dose-dependently attenuated anti-CD3- and anti-CD28-activated human PBMNC proliferation compared to nonactivated PB-MNCs. ap < 0.05 versus control; bp < 0.05 versus vehicle. n = 3–4/group. (A, F, H) ANOVA with post hoc Newman–Keuls test; (C) Student's t-test.
To determine the effect of ADSCs on lymphocyte proliferation, we added either ADSC- or TNF-α-treated ADSC conditioned medium (ADSC-CM) to an hPBMNC culture in the presence or absence of anti-CD3 and CD28 antibodies 47 . ADSCs as well as ASC-CM markedly inhibited lymphocyte proliferation in a dose-dependent manner (Fig. 3H).
Collectively, studies present ADSCs as pleiotropic immunomodulators of macrophages, neutrophils, and lymphocytes, in a cell contact-independent manner. In the context of AAA, this suggests that the presence of ADSCs may limit cytotoxic lymphocyte activation and the neutrophil infiltration 48 associated with the development of abdominal aortic aneurysm in vivo.
Human ADSCs Suppress Elastase-Induced AAA Expansion
Previous studies have revealed that the vasa vasorum of the aortic tunica adventitia layer represent an important source of infiltrating inflammatory cells2,49–52. The chronic nature of this inflammation initiates and drives the progressive degradation of aortic collagen along with extracellular matrix elastin fragmentation 53 . To simulate this abluminal mechanism of injury, we induced murine aortic inflammation by applying topical elastase to the aortic adventitia in absorbable gelatin foam. As anticipated and observed in human AAA specimens, the adventitial presence of elastase led to massive infiltration of inflammatory cells by day 7 (Fig. 4). The severity of this aortic inflammation was confirmed by the progressive expansion of abdominal aorta as seen through ultrasound (Fig. 5A) and videomicrometry (Fig. 1). Remarkably, a single injection of hADSCs significantly suppressed this expansion as early as day 4 [aortic diameter increase from baseline: 54.8% (saline) vs. 16.9% (ADSCs), n = 4/group, p < 0.001]. Such therapeutic effect of hADSCs persisted through the end of the study period on day 14 (aortic diameter increase from baseline 64.1% saline vs. 24.6% ASDCs, n = 4/group, p < 0.01) (Fig. 5B). Overall, these results demonstrated that a single IV injection of hADSCs was sufficient to limit aortic diameter expansion following AAA initiation in this model.
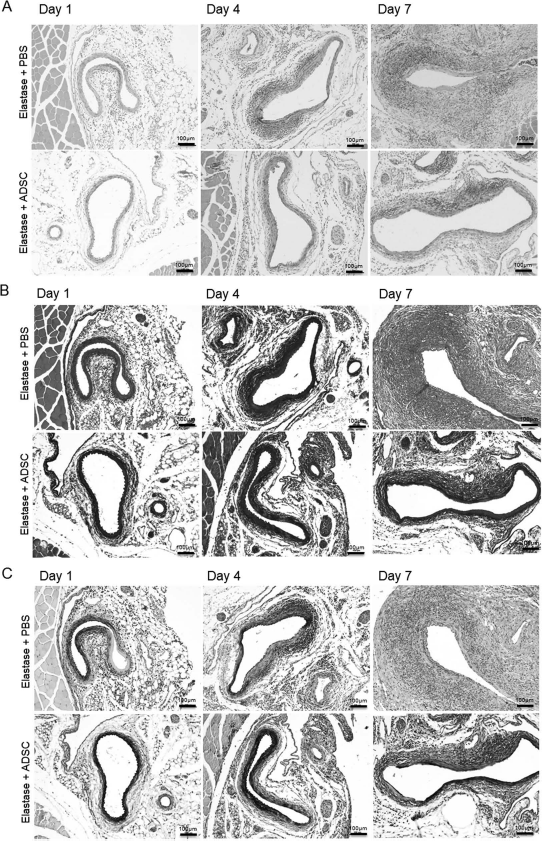
Human ADSC treatment results in reduced periaortic inflammation, and greater preservation of elastin and collagen within the aortic wall in an elastase-induced murine abdominal aortic aneurysm (AAA) model. Representative (A) H&E, (B) elastin, and (C) Masson's trichrome staining of ADSC- and control (PBS)-treated abdominal mouse aneurysms on days 1, 4, and 7. Scale bars: 100 μm.

Human ADSCs suppress elastase-induced AAA expansion in C57BL/6 mice. (A) Mouse aortic ultrasound images at baseline and days 4, 7, and 14 post-elastase treatment, in conjunction with administration of either vehicle or 1 × 106 human ADSCs [intravenous (IV)]. (B) ADSCs significantly suppressed murine elastase-induced aortic expansion starting from day 4 and persistently until day 14, compared to vehicle (n = 4–10/group, ∗∗p < 0.01 Student's t-test).
Human ADSCs Induce a Rapid Aortic Surge in Foxp3+ Tregs with a Corresponding Decline in Inflammatory CD28− T Cells and Neutrophils
AAA patients experience a deficiency in aortic Tregs with an accompanying prevalence of CD28− T cells and neutrophils27,28. We determined that these features were present in our murine model of AAA and were reversible in the presence of ADSCs. Consistent with our in vitro studies, ADSCs induced a rapid and significant 20-fold surge in aortic Tregs (Fig. 6A and B) compared to controls. This was associated with a marked repression of the total aortic T-cell population by 58% and 75% at days 1 and 4, respectively (Fig. 6C). Notably, the initial inductive effect of ADSCs on Treg was weakened over 7 days after the initial cell delivery, which is consistent with loss of suppression on total T cells at day 7 by ADSCs, likely reflecting a diminishing effect of ADSCs 7 days after the initial dose of injection. Other inflammatory cell populations demonstrated responses to ADSC injection similar to those found for the total T-cell population. At day 1, the CD4+CD28− and CD8+CD28− T-cell subsets were already significantly decreased by 28% and 61%, respectively (Fig. 7A). This was accompanied by a 43% decrease in Ly6G/C+ neutrophil infiltration (Fig. 7B and C). The early ADSC-induced increase in the AAA aortic Treg content with concurrent inhibition of CD28− T-cell and neutrophil infiltration likely contributed to the observed day 4 suppression of aortic expansion.

Human ADSCs increased the abdominal aortic ratio of Foxp3+ Tregs to CD3+ T cells in our model of AAA. (A) Representative immunohistochemical (IHC) staining of CD3 (total T cells) and FoxP3 (Tregs) for murine AAA on day 7. (B) Total CD3+ and FoxP3+ cells in the entire tunica adventitia and media layers were counted by microscopy of entire aortic sections at the point of maximal aortic circumference. The percentages of Tregs among total T cells were persistently increased by ADSCs throughout the first 7 days after cell injection. (C) Quantification of the density of total T cells showed suppression of T-cell infiltration by ADSCs in the early stage of AAA development. Arrows indicate positively stained cells (n = 4–5/group, ∗p < 0.05, ∗∗p < 0.01, Student's t-test).

Human ADSCs significantly reduce inflammatory T-cell and neutrophil populations in elastase-induced abdominal aortic aneurysm. Flow cytometry was gate for (A) CD4+CD28− and CD8+CD28− T cells, and (B) neutrophils (Ly6G/C+). (C) ADSCs decreased inflammatory subset levels compared to controls (n = 4–5/group, ∗p <0.05, ∗∗p <0.01, Student's t-test).
Human ADSCs Reduce Circulating Proinflammatory Monocytes and Polarize Aortic Macrophages Toward the M2 Subtype
Systemic circulation contributes to the initial AAA inflammatory response by delivering monocytes to the site of aortic inflammation 16 with proinflammatory M1 macrophages defining the majority of the AAA-stimulated inflammatory cell infiltration 54 . In our topical elastase-induced AAA model, we observed that the presence of ADSCs progressively reduced the circulating levels of proinflammatory CD115+CXCR1−LY6C+ monocytes, with significance achieved by day 7 (Fig. 8A and B). The loss of significance between the two treatment groups by day 14 is likely a consequence of resolving systemic inflammatory response to the initial elastase insult in the context of a small sample size. This reduction in circulating monocytes was associated with aortic resident macrophage polarization toward the reparative M2 subtype (CD206+) (Fig. 8C–E). Collectively, these results suggest that early ADSC intervention may be a clinically relevant therapeutic means of modulating the initial inflammatory components linked to exacerbation of abdominal aortic aneurysm.

Human ADSCs reduced circulating proinflammatory monocytes and polarized aortic macrophages to the M2 subtype. (A, B) ADSCs significantly decrease the ratio of inflammatory monocytes (CD115+Ly6C+CX3CR1−) to resident monocytes (CD115+Ly6C−CX3CR1+) by day 7 in AAA mouse peripheral blood, compared to control. (C) Day 7 AAA IHC staining for CD68 and CD206. Scale bar: 50 μm. (D, E) ADSCs provided a significant increase in the day 7 CD206+/CD68+ (M2/total) macrophage ratio, with a corresponding day 7 decrease in total (CD68+) macrophages. (B) n = 6–10/group, ∗p < 0.05 (D, E) n = 3–5/group. ∗p < 0.05 versus PBS group, Student's t-test.
Intravenous ADSC Administration Leads to Transient Lung Trapping Without Aortic Engraftment
Hoechst-labeled ADSCs were injected into the tail vein of control and AAA-induced mice. Consistent with other observations46,55, the biodistribution of the 1 × 106 ADSCs that were injected exhibited predominant pulmonary localization on day 1 with no engraftment at the site of aortic injury (Fig. 9A). ADSC lung entrapment measured at 24 and 96 h postinjection demonstrated a time-dependent decrease in concentration. ADSC entrapment in lungs was quantified using a standard curve of in vitro mixed mouse lung tissue titrated with hADSCs. This curve could detect ∼1,000 cells/bilateral lungs or as low as 0.01% of the delivered IV dose (Fig. 9B). Quantitatively, an average of 3% (up to 15% in some individuals) of the delivered cells were retrievable from the lungs by 24 h, whereas less than 0.5% could be retrieved by 48 h (Fig. 9C).

Intravenously administered ADSCs were not found frequently in the aorta and were transiently trapped in lung tissue. (A) Fluorescence imaging of lung (top) and aorta (bottom) showing presence of Hoechst-labeled human ADSCs on days 1 and 4. Scale bar: 50 μm. (B) Human Alu polymerase chain reaction (PCR) sequence curve showing a linear correlation between Ct value and human ADSC number in the mouse lung. (C) Less than 1% of human-infused ADSCs were detected in the day 4 mouse (n = 5–8/group).
Discussion
To date, no reports on the effect of ADSCs on neutrophil transmigration have been published, and there are limited studies on M2 macrophage 56 and Treg36,37 induction. Moreover, none have demonstrated the potential involvement of ADSC-mediated paracrine factors in these activities. Our results suggest that ADSC paracrine signaling may be a useful immunomodulatory element in the regulation of AAA expansion. Using both in vitro and in vivo experiments, we demonstrated that ADSCs can positively regulate macrophage polarization toward the M2 reparative phenotype, inhibit neutrophil transmigration, and induce the aortic expression of immunosuppressive Foxp3+ Tregs in a murine model of AAA. Consistent with the literature38,57, the presence of ADSCs after aneurysms reduced the level of circulating levels of proinflammatory CD115+CXCR1−LY6C+ monocytes.
In testing the influence of ADSC conditioned media on cellular inflammatory characteristics, we found that, indeed, ADSC conditioned media (ADSC-CM) generated from TNF-α-stimulated ADSCs can suppress CD3/CD28 Ab-induced lymphocyte proliferation compared to conditioned medium from unstimulated ADSCs. The TNF-α-stimulated ADSC-CM was able to dose-dependently inhibit lymphocyte proliferation, indicating that the cellular environment has a significant role in determining ADSC immunomodulatory activities. Taken together, these results robustly support the idea that ADSC-mediated paracrine factors exert a therapeutic, immunomodulatory effect in aortic inflammation.
AAA development and expansion are relatively rapid in models of AAA compared to the chronic clinical course of aneurysm development in human patients 58 . This dichotomy raises the question of how accurately AAA models may predict human clinical disease progression and outcomes 1 . Despite this, each model offers mechanistic and therapeutic insight into key pathological components associated with AAA development and progression. Given the importance of the adventitial layer and its vasa vasorum in mediating inflammatory cell infiltration49,59,60, we selected a topical elastase-based approach to experimentally induce aortic inflammation and subsequent expansion 61 . The resulting inflammatory cell infiltration from the vasa vasorum has been shown to mimic observations in human aneurysms62–65. We further optimized this approach using Gelfoam-mediated periadventitial elastase application to specifically focus on early adventitial inflammation. Our objective was to determine if the anti-inflammatory effects of ADSCs could limit the inflammatory initiation and early growth of small aneurysms. We determined that topical elastase potently stimulated and sustained abdominal aortic aneurysm formation. This method has the advantage of avoiding aortic mechanical or needle injuries with a complimentary reduction in postoperative complications (<10%) and mortality (<5%). This rapid and reproducible method generated an aorta-specific inflammatory environment in which the sequential modulation of cellular and peripheral inflammatory events could be studied in response to ADSC administration.
We found that aortic topical elastase application produced massive inflammatory cell infiltration in the vasa vasorum as well as degradation of collagen and elastin fibers by day 7 (Fig. 4). In monitoring the expected sequential aortic changes, we focused on the early, first week, time points as opposed to previous chronic studies31,34,66. Our rationale is that a detailed temporal delineation of the early AAA inflammatory 27 events may lead to therapeutic innovations to limit aneurysmal progression at early stages. We demonstrated that IV injection of 1 × 106 hADSCs at the time of early aneurysm formation generated a rapid surge in Tregs on day 1 with a corresponding decline in CD28− T cells. This, in conjunction with the subsequently observed (day 4) suppression of aortic expansion, suggests a central role of early aortic inflammation in the pathogenesis of aneurysm development. This novel observation provided us an avenue by which to mechanistically study the involvement of CD28− T cells and Tregs in the development of AAA.
Recent evidence has also elucidated a role for monocytes in the evolution of large aneurysms 40 . Our endpoint analysis found that the presence of hADSCs after AAA decreased the levels of circulating monocytes. ADSC administration significantly decreased the circulating levels of CD115+Ly6C+CX3CR1− inflammatory monocytes compared to CD115+Ly6C−CX3CR1+ resident monocytes (Fig. 7). This was further supported by the parallel increase in aortic anti-inflammatory M2 macrophages by day 7 (Fig. 8). The contribution of M2 macrophages to the amelioration of AAA has only recently come to light through the modulation of peroxisome proliferator-activated receptor-γ (PPAR-γ) and Notch signaling inhibition67,68. We are the first to demonstrate that ADSCs may influence the activity of M2 macrophages in the context of AAA. Our consistent observation of ADSC-mediated M2 macrophage upregulation and complementary Treg production with the terminal suppression of aneurysm expansion presents immunomodulation as an important therapeutic approach to limiting AAA progression.
To ascertain ADSC biodistribution in the context of AAA therapy, we measured ADSC aortic engraftment and lung entrapment. Consistent with other animal models69–73, we found that IV ADSC delivery resulted in the distribution of ADSCs predominantly in the lungs with minimal aortic engraftment (Fig. 9A). Consistent with observations in previous studies30–35, our findings highlighted the therapeutic importance of ADSC paracrine factors and paracrine signaling as a major component of their regenerative capacity. Moreover, we found the presence of ADSCs in the lungs to be transiently cleared by day 4. This was consistent with a previous study wherein researchers assessed lung clearance of bone marrow-derived MSCs (BM-MSCs) in mice with myocardial infarction and showed that <0.1% of infused cells remained in the lungs after 96 h 43 . This acute cell clearance in contrast to the chronic suppression of aortic expansion over the course of 2 weeks strongly supports the idea that an ADSC-mediated paracrine effect was therapeutically important. The local physical presence or aortic engraftment of ADSCs appears unnecessary to perpetuate changes in the aneurysm models of an inflammatory environment and suppresses AAA progression. A single ADSC dose of 1 × 106 was sufficient to modulate the AAA immune response and to upregulate Treg and M2 macrophage expression in this model. Concordantly, ADSCs downregulated monocyte activation, neutrophil transmigration, and circulating CD28− T-cell levels, thereby inhibiting aneurysm progression. Overall, our results call for future experiments examining the individual inflammatory components contributing to the therapeutic effects of ADSCs for AAA.
Conclusion
A single IV injection of 1 × 106 ADSCs is sufficient to suppress abdominal aortic expansion in a topical elastase-induced murine model of AAA. Periadventitial application of elastase-soaked Gelfoam successfully induced AAA formation in mice and demonstrated an orchestrated activation of inflammatory monocytes, Tregs, and macrophages, which is consistent with human AAA patients. The therapeutic effect was experimentally attributed to the early and immediate ADSC-mediated suppression of neutrophil transmigration and circulating lymphocyte activation following AAA. Importantly, the regenerative potential of ADSCs was observed through the aortic induction of M2 macrophages and Tregs. The infused ADSCs were predominantly localized to the lungs and did not engraft in the aortas of AAA models. This observation supports our hypothesis that ADSC-mediated paracrine factors modulate the AAA-induced inflammatory response to suppress aneurysm expansion and AAA progression.
AAA is a lethal disease with chronic progression leading to end-stage aneurysm rupture and an 80% mortality rate. Currently, AAA treatment options include open surgical and endovascular repair preceding rupture and are associated with significant morbidity, mortality, and expense. In this article, we present an early and proactive way to mediate and suppress aortic expansion through a single abdominal aortic stem cell infusion. The human ADSCs were readily accessed through minimally invasive liposuction procedures and isolated in large quantities, offering a novel and practical modality for autologous AAA treatment. AAA was induced by a single elastase-containing gel foam application to the abdominal aorta in mice. Considerable dynamic and concerted systemic and aortic inflammatory cellular events were observed in the early stage of murine AAA development. This is consistent with what has been previously observed in AAA patients, but not reproduced or reported in animal models.
Importantly, these results demonstrate that ADSC infusion, in our AAA model, produced an immunomodulatory shift in the animal and aortic tissue from a predominantly proinflammatory state to a tissue-reparative, anti-inflammatory state. This was observed through decreases in CD28− T cells, activated monocytes, neutrophils, and M1 macrophages, with corresponding increases in tissue-reparative Tregs and M2 macrophages. We propose that this feed-forward anti-inflammatory cascade prompted the suppression of aortic expansion and aneurysm development.
Notably, results derived from both in vivo biodistribution studies and in vitro cell culture studies demonstrate that ADSC paracrine factors mediate the AAA anti-inflammatory activity. This preclinical evidence of ADSC efficacy in the amelioration of early AAA inflammation and chronic expansion provides strong evidence for future studies and clinical trials using autologous ADSCs or ADSC-derived cellular products in the treatment of early stage small AAA.
Footnotes
Acknowledgments
This work was supported by the IU Health Center for Aortic Disease, the Indiana Center for Vascular Biology and Medicine, and the Richard L. Roudebush Veterans Affairs Center for Regenerative Medicine. Support also comes from Merit Review Award (#CX000263) from the United States Department of Veterans Affairs and T32 Award (NHLBI 05999) from National Institutes of Health. Thanks are due to Dmitry Traktuev for valuable comments on the experiments and the manuscript. The authors declare no conflicts of interest.