
Abstract
Hair follicle stem cells (HFSCs) are considered one of the useful donor cell types for skin regenerative medicine owing to their robust proliferative capacity and multipotency. However, methods for easily and effectively obtaining HFSCs from a limited skin biopsy are still lacking. Here we report a novel approach for obtaining a subpopulation of HFSCs from a small skin sample from the rat tail, which uses the sebaceous glands (SGs) to capture the adjacent HFSCs. By means of organ culture, keratinocytes were expanded from the detached SGs, which also included adherent HFSCs from the hair follicle that could be passaged at the single-cell level. These SG-captured keratinocytes strongly expressed the basal layer markers K14, integrin α6, and p63; the bulge stem cell marker K15; and the upper isthmus stem cell marker Plet1. Furthermore, we reconstituted new epidermis, hair follicles, and SGs from the SG-captured keratinocytes using an easily operated, modified skin reconstitution assay based on silicone gel sheeting. This study suggests that the SGs could be an accessible capturer to harvest the adjacent HFSC subpopulation, particularly when the donor tissue is limited.
Introduction
Skin is an important barrier for animals, especially mammals. As the largest tissue, skin, which is composed of the epidermis and dermis, can protect bodies from multiple conditions, such as water loss, light irradiation, and stress 1 . In haired mammals, the hair follicle, a miniorgan constructed from multilayered epithelial keratinocytes, is a significant component of the epidermis2,3. Because organ homeostasis relies on the presence of stem cells, hair follicle stem cells (HFSCs) are considered to be responsible for hair follicle homeostasis, which is essential for the physiological functions of the skin4–6. HFSCs have the capacity to form colonies and undergo self-renewal and multidifferentiation, which are properties shared by many adult stem cells7–10. On the one hand, HFSCs provide undifferentiated daughter cells, which are retained in the similar niche and hold the same characteristics of the parent cells. On the other hand, differentiation-committed daughter cells are also born at the same time, and these cells subsequently differentiate into the keratinocytes that constitute hair follicles, which undergo periodic growth and degradation cycles during the life span of mammals 11 . In addition to the normal physiological role of HFSCs in vivo, HFSCs can participate in the wound repair process when a wound is deep enough in the skin12,13. Moreover, HFSCs could be used as a novel source of multipotent stem cells for tissue engineering and regenerative medicine14,15.
HFSCs are found within at different regions of the hair follicle outer root sheath (ORS) 7 , which can be defined as the elongation and fold of the basal layer of the epidermis that is the location of interfollicular epidermal stem cells 16 . Therefore, HFSCs residing in the ORS express the epidermal stem cell markers K5/K14 9 , p63 17 , and integrin α6 18 . In addition, HFSCs express specific markers, such as CD34 10 and K15 9 ; however, there are several HFSC subpopulations residing along the ORS19,20 that have distinct gene expression profiles. For example, infundibulum stem cells express stem cell antigen 1 (Sca1) positive 21 ; upper isthmus stem cells express placenta-expressed transcript 1 (Plet1)/MTS24 22 and leucine-rich repeats and immunoglobulin-like domain protein 1 (Lrig1) 23 ; upper bulge stem cells express leucine-rich repeat-containing G protein-coupled receptor 6 (Lgr6) 24 ; and stem cells residing in the bulge, which are the earliest discovered HFSC reservoir 25 , express CD34 10 and leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5) 26 . Despite the existence of differences between these HFSC subpopulations, they all serve the homeostasis and functions of the epidermis.
The traditional methods for obtaining HFSCs are fluorescence-activated cell sorting (FACS) and microdissection. Compared with FACS, which requires the expression of specific cell surface markers and a large amount of skin tissue, microdissection and organ culture have advantages, such as an easy operation and a relative need for small amounts of skin tissue27,28. For clinical applications, obtaining HFSCs efficiently from limited skin biopsies remains a challenge. We previously demonstrated that abundant HFSCs could be obtained from a single rat or ovine vibrissa hair follicle via microdissection and organ culture29,30. Nonetheless, this microdissection method does not work as well for small or fragile hair follicles, such as those in the skin from the back and tail.
In this study, we further developed a more feasible method for acquiring HFSCs from the upper isthmus (a part of the ORS next to the opening of SGs) of the tail hair follicles, using isolated sebaceous glands (SGs) as the capturer. Moreover, we were able to reconstitute epidermal structures in the back skin of the nude mice using these HFSCs and neonatal dermal cells in a modified skin reconstitution model. This technique only requires the isolation of the superficial part of hair follicles rather than an intact hair follicle. Furthermore, this method is suitable for various types of hair follicles, making it possible to obtain HFSCs from different and limited skin tissues. The present study helps to overcome the limitations associated with HFSC sources and enhances the potential applications of HFSCs in clinical wound repair and skin tissue engineering.
Materials and Methods
Animals
The Brown Norway (BN) rats (10 weeks) and athymic nu/nu nude mice (7–8 weeks) used in this study were purchased from Vital River Laboratories Co. Ltd. (Beijing, P.R. China). The mice were fed in the animal facility of the Institute of Zoology, Chinese Academy of Sciences (CAS), were maintained in 12-h light/12-h dark conditions, and had ad libitum access to food and water. The experiments were conducted in accordance with the consent of the ethics committee of the Institute of Zoology, CAS (under the project identification code, the Strategic Priority Research Program of the CAS XDA01010202, 201101-201512). The experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals issued by the Institute of Zoology, CAS.
Feeder Cell Preparation
Neonatal rats were disinfected by multiple washes with saline, and their skin was obtained. Then the skin was floated on 5 mg/ml dispase (Gibco, Carlsbad, CA, USA) at 4°C overnight to separate the epidermis and dermis. The next day, the dermis was digested with 10 mg/ml type IV collagenase (Gibco) into single cells that were then seeded at a density of 2 × 106 cells per 10-cm dish and cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) and penicillin–streptomycin (Sigma-Aldrich, St. Louis, MO, USA). Before their use as feeder cells, P3 neonatal rat dermal fibroblasts were pretreated with 10 μg/ml mitomycin C (Sigma-Aldrich).
SG Isolation and Culture
Tail skin (approximately 1 cm × 1 cm) was collected from BN rats. After disinfecting with povidone–iodine (Lerkam Biotechnology Co., Ltd., Hunan, P.R. China), 75% (v/v) ethanol (Beijing Chemical Works, Beijing, P.R. China), and sterile saline (Shandong Hualu Pharmaceutical Co. Ltd., Shandong, P.R. China), the skin was placed into a 6-cm dish (Corning, Corning, NY, USA) containing a 5-mg/ml dispase solution (Gibco) for 1 h at 4°C followed by 30 min at 37°C. After the epidermis was removed from the skin, the residual dermis was transferred into clean phosphate-buffered saline (PBS) and shaken gently; the SGs were then observed to have detached and to float in the solution. These SGs were then pipetted into 12-well plates (Corning) at 37°C/5% CO2 in a growth medium consisting of William's E medium (Gibco) containing 15% FBS (Hyclone, Logan, UT, USA), 10 ng/ml epidermal growth factor (EGF), 5 μg/ml insulin, 1 μg/ml hydrocortisone, 10 μg/ml transferrin, 0.1 μg/ml linoleic acid–bovine serum albumin (BSA), 0.115 μg/ml vitamin A, 1 μg/ml vitamin D2, 1× glutamine, and 1× penicillin and streptomycin (all from Sigma-Aldrich), as described previously10,29–31. The growth medium was changed every second day.
The workflow of this study is illustrated in Figure 1.
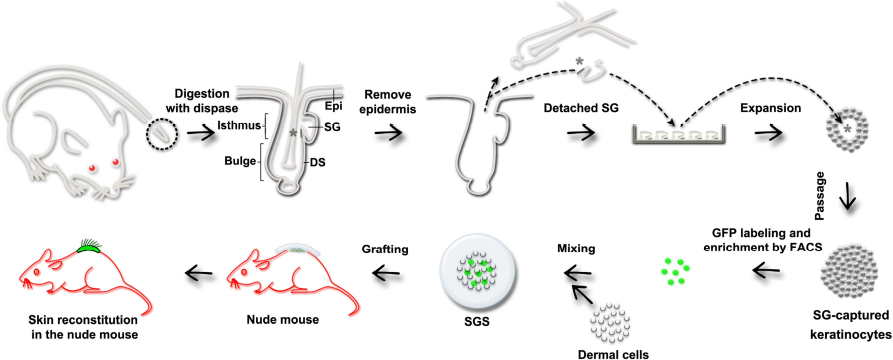
Schematic diagram of the experimental design. Along the direction of the arrows, skin obtained from a small tail biopsy was digested with dispase solution to remove the epidermis and detach the SG from the corresponding dermis. The SGs were pipetted into a petri dish containing culture medium, and after purification and expansion, a large number of HFSCs were harvested. To track the cells during skin reconstitution, the HFSCs were labeled by GFP-expressing lentivirus and grafted into the back of nude mice along with neonatal rat dermal cells. Asterisks indicate the HFSCs in the isthmus region close the SG in the hair follicle. SG, sebaceous gland; DS, dermal sheath; SGS, silicone gel sheeting.
Keratinocyte Purification and Passaging
On the basis of their adherent properties, HFSCs can be easily purified by a two-step trypsinization procedure.
Non-HFSCs and residual SGs were detached from the plate by treatment with 0.1% trypsin–0.008% ethylenediaminetetraacetic acid (EDTA) (Gibco) for 3–5 min at room temperature (RT), while the HFSCs remained on the plate. After rinsing with PBS, the HFSCs can be further dissociated with 0.25% trypsin–0.02% EDTA (Gibco) for approximately 5 min at 37°C. Following the inactivation of trypsin by FBS, a single-cell suspension of HFSCs was plated with neonatal rat fibroblasts as the feeder cells (1:3, HFSC/feeder cells). The growth medium was changed every second day, and the cells were passaged at least to P10.
For single-cell passaging, the SG-captured keratinocyte suspension was diluted to a very low concentration, and under a microscope (Nikon, Tokyo, Japan), single cells were pipetted into one well of a 48-well plate (Corning) with feeder cells and cultured using the above method.
RNA Extraction, Reverse Transcription-PCR (RT-PCR), and Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from P5 and P10 HFSCs and rat dermal fibroblasts using a TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. After the removal of genomic DNA with RQ1 RNase-Free DNase (Promega, Madison, WI, USA), Moloney murine leukemia virus (M-MuLV) reverse transcriptase (New England Biolabs, Beverly, MA, USA) was used for reverse transcription. RT-PCR was performed in a 20-μl reaction volume with Promega PCR Master Mix (Promega) as described previously 32 , and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used for normalization. RT-PCR was performed using the following settings: 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, for 30 cycles. The primers used for the RT-PCR are listed in Table 1.
Primer Sequences Used for RT-PCR

qRT-PCR was performed using a Roche LightCycler 480 system (Roche Applied Science, Indianapolis, IN, USA) and the GoTaq qRT-PCR master mix (Promega) with a preincubation at 95°C for 2 min; followed by 40 cycles of amplification at 95°C for 15 s, 60°C for 15 s, and 70°C for 15 s. β-Actin was used for normalization, and the primers used for the qRT-PCR are listed in Table 2.
Primer Sequences Used for qRT-PCR

Rhodamine B Staining and Immunofluorescence
SG-captured keratinocytes were seeded onto coverslips, cocultured with fibroblasts for approximately 4 days to form colonies, and then fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich) for 10 min at RT and rinsed three times with PBS. For Rhodamine B staining, colonies were stained with 1% Rhodamine B (Sigma-Aldrich) and then photographed with a Canon G12 camera (Canon, Tokyo, Japan). For immunofluorescence, if necessary, 0.05% Triton X-100 (Sigma-Aldrich) was used for permeation. Nonspecific binding was blocked in PBS containing 5% BSA (Sigma-Aldrich) for 1 h at 37°C. Cells were incubated in a humid chamber at 4°C overnight with anti-K15 (Santa Cruz Biotechnology, Dallas, TX, USA), anti-p63 (Santa Cruz Biotechnology), anti-K14 (EMD Millipore, Billerica, MA, USA), and anti-integrin α6 antibodies (AbD Serotec, Raleigh, NC, USA). After three washes in PBS, the cells were incubated with the corresponding secondary antibodies (rabbit anti-goat secondary antibody for anti-K15 primary antibody; goat anti-rabbit secondary antibody for anti-p63 primary antibody; goat anti-mouse secondary antibody for anti-K14 primary antibody; and goat anti-rat secondary antibody for anti-integrin α6 primary antibody; all dilutions were 1:100, and all secondary antibodies were from Zhongshan Goldenbridge Biotechnology Co. Ltd., Beijing, P.R. China) diluted in blocking solution for 1 h at 37°C, followed by nuclear counterstaining with propidium iodide (PI) (Sigma-Aldrich) for 10 min at RT. After three rinses with PBS, the cells were mounted with 1,4-diazabicyclooctane (DABCO) (Sigma-Aldrich). The cells were observed and photographed using Leica TCS SP2 confocal microscope (Leica Instruments, Nussloch, Germany).
Lentivirus Production and Infection
Lentivirus production was performed as previously described 33 . Briefly, the lentiviral vectors for infection were packaged using ViraPower™ Lentivirus Packaging System (Invitrogen) according to the manufacturer's instructions. 293FT cells (Invitrogen) at nearly 50% confluence were transfected with target plasmids for 2K7-green fluorescent protein (GFP)/Neo and supermix plasmids (Invitrogen) using the Fugene HD transfection reagent (Roche Applied Science). After an overnight incubation, the medium was replaced, and after 48 and 72 h supernatant containing lentivirus was collected and concentrated with ultracentrifuge (Beckman Coulter, Brea, CA, USA) at 48,400 × g for 1.5 h at 4°C.
For infection, 105 SG-captured keratinocytes were incubated for 24 h in growth medium containing concentrated lentivirus and 8 μg/ml polybrene (Sigma-Aldrich). The virus-containing growth medium was replaced with fresh growth medium after 24 h. Highly fluorescent, GFP-labeled SG-captured keratinocytes were enriched by a BD Aria flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and cultured as previously described.
Skin Reconstitution and Histological Analysis
Neonatal dermal cells were prepared as described previously 34 . Briefly, the trunk skin of neonatal BN rats was mechanically separated and floated in 5 mg/ml dispase (Gibco) overnight at 4°C. After discarding the epidermis, the remaining dermis was cut into pieces, digested in 10 mg/ml type IV collagenase (Gibco) for 40 min at 37°C, and further centrifuged several times to obtain a single-cell suspension. The GFP-labeled SG-captured keratinocyte single-cell suspension (P7) was prepared with the previous methods. Then the dermal cells were mixed with GFP-labeled SG-captured keratinocytes at a ratio of 5:1 (1.8 × 107 dermal cells: 3.6 × 106 keratinocytes) in 150 μl of DMEM (Gibco). The cell mixture plus 50 μl of Matrigel (Becton Dickinson) was allowed to adhere to the sticky surface of the auxiliary scaffold, which consisted of Epi-Derm® Silicone Gel Sheeting (SGS) (Biodermis, Henderson, NV, USA) at a diameter of 3 cm, for 2 h at 37°C. When the cell mixture had adhered to the sheeting, it was placed onto a wound in the back skin of an athymic nude mouse. The dorsal skin of the mice was marked with a 1.5-cm-diameter circular stamp, and the full skin was cut off using scissors. The cell mixture was fixed precisely in the wound by the covering SGS (Biodermis). After 4 weeks, when new hair began growing from the skin graft, the mice were sacrificed, and the graft sites were cryosectioned into 40-μm-thick slices using the CM1950 platform (Leica). After fixing in 4% PFA, the sections were counterstained with PI (Sigma-Aldrich) and further observed and photographed under a Zeiss LSM710 confocal light microscope (Carl Zeiss, Jenna, Germany).
Statistical Analysis
Data are shown as the means ± SEM. Data were analyzed by one-way analysis of variance (ANOVA) and a Bonferroni post hoc test or by an independent t-test with SPSS 16.0 (IBM, Amronk, NY, USA). A value of p < 0.05 was considered to be statistically significant.
Results
Isolation and Purification of SG-Captured Keratinocytes
The tail skin, which contains numerous hair follicles, has been considered to be an outstanding model for studying the morphology and function of mammalian pilosebaceous unit35-37. For example, there are approximately 30 hair follicles arranged in triplets within a 9-mm2 area of rat tail skin (Fig. 2A), which was a sufficient number to complete the following study. According to our observations and the existing literature36,37, tail hair follicles in the relatively uniform anagen have prominent and hypertrophic SGs, which is a considerable advantage for obtaining neighboring HFSCs. After the epidermis was removed from the digested skin, the SGs were able to be easily collected using a pipette. Under a stereoscope and inverted microscope, we observed intact structures consisting of separated SGs and affiliated cell layers (Fig. 2B and C). According to the hair follicle architecture, these accessory cell layers were considered to be the upper isthmus, a part of the ORS next to the opening of SGs. After 2 days of primary organ culture, keratinocytes grew from the adherent tissue (Fig. 2D). Subsequently, these keratinocytes were passaged over a considerable time with stable and homogeneous morphology in vitro. For instance, at passage 10 (P10), these cells exhibited the classic cobblestone morphology 3 days after passage (Fig. 2E and F). Thus, we named these cells SG-captured keratinocytes.

Isolation of keratinocytes from a small biopsy of rat tail skin. (A) Dorsal view of the tail skin tissue after digestion. (B, C) Observation of the detached SG and accessory cell layers as viewed under a stereoscope (B) and an inverted microscope (C). Asterisk indicates the accessories from the upper isthmus of the outer root sheath. (D) Keratinocytes growing out from the adherent tissue. (E, F) Keratinocytes after long-term passage (P10) possessing a typical cobblestone morphology. (G) Colony formed from a single cell stained by Rhodamine B. SG, sebaceous gland; Fb, fibroblast. Scale bars: 200 μm (B–F), 5 mm (G).
In a single-cell cloning assay, P9 keratinocytes (45.83 ± 2.08%) formed compact colonies with a clear edge as shown by staining for Rhodamine B (Fig. 2G). Beginning with a single cell seeded on day 0, the final cell number on day 10 reached 6.36 ± 1.32 × 104 cells, indicating the strong proliferative capacity of these SG-captured keratinocytes and the high yield of this method. Thus, the ability to long-term passage and the high proliferative capacity of these SG-captured keratinocytes in vitro suggested that these cells might be a type of stem cell from the upper isthmus.
Characterization of SG-Captured Keratinocytes
To determine the characteristics of these long-term passaged SG-captured keratinocytes, we first investigated the expression of epidermal stem cell-related markers, especially those for HFSCs, by quantitative and qualitative RT-PCR. On the basis of our results, SG-captured keratinocytes at P5 and P10 expressed the basal layer marker keratin 14 (Krt14), the epithelial stem cell markers integrin α6 (Itga6) and p63 (Trp63), the upper isthmus stem cell marker Plet1, and the bulge stem cell marker keratin 15 (Krt15) (Fig. 3A). As negative controls, rat dermal fibroblasts expressed none of these markers (Fig. 3A). However, the expression levels of another upper isthmus stem cell marker Lrig1 and the bulge stem cell marker Cd34 in SG-captured keratinocytes were weak, and the expression of the upper bulge stem cell marker Lgr6 was not detected (Fig. 3B). Regarding the lack of Cd34 and Lgr6 expressions in these SG-captured keratinocytes, it is possible that there are species variations across lab rodents or that the expression profiles in vivo and in vitro are different. In contrast to the feeder cells, immunofluorescence staining further indicated the specific expression of keratin 14 (K14), integrin α6, p63, and keratin 15 (K15) in SG-captured keratinocyte colonies (outlined areas in Fig. 3C). These results suggested that SG-captured keratinocytes showed the gene expression signatures of HFSCs in the isthmus.
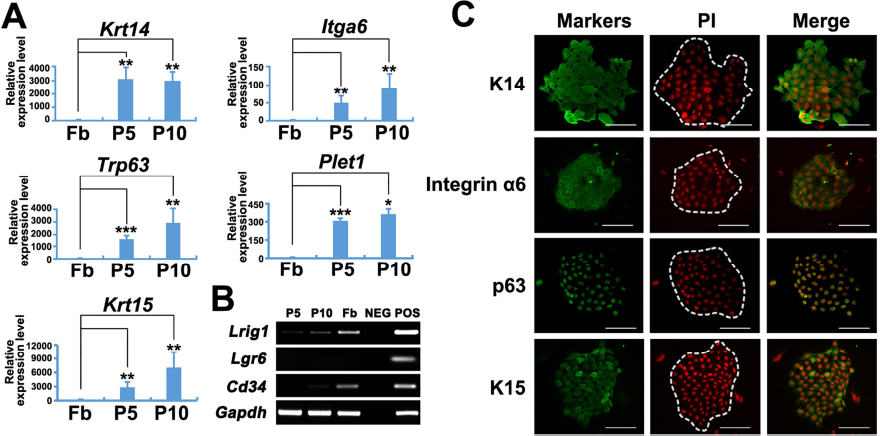
Characterization of SG-captured keratinocytes. (A) qRT-PCR results showing the mRNA expression of Krt14, Itga6, Trp63, Plet1, and Krt15 in P5 and P10 SG-captured keratinocytes. Rat fibroblasts served as a negative control. (B) RT-PCR results showing the expression of Lrig1, Lgr6, and Cd34 in P5 and P10 SG-captured keratinocytes. H2O served as the negative control; neonatal rat epidermis served as the positive control. (C) Immunofluorescence staining for the specific expression of K14, integrin α6, p63, and K15 in colonies formed by SG-captured keratinocytes (the outlined areas). Fb, fibroblast; P5, passage 5; P10, passage 10. Scale bars: 100 μm.
Skin Reconstitution Using SG-Captured Keratinocytes in a Modified Skin Reconstitution Model
For HFSCs, multipotency was demonstrated by their ability to differentiate into epidermal lineage cells, such as the cells in hair follicles and interfollicular epidermis. Thus, the reconstitution of new hair follicles and epidermis in the back skin of nude mice is considered to be an essential feature of HFSCs. To investigate the multipotency of SG-captured keratinocytes, we performed a modified skin reconstitution assay with these cells and neonatal rat dermal cells in nude mice. For subsequent observations, we labeled SG-captured keratinocytes with GFP by lentiviral infection and purified the labeled cells by FACS. The proportion of the strongly GFP-positive SG-captured keratinocytes was 9.5%, and these GFP-labeled cells retained their colony formation ability (Fig. 4A). As in other reconstitution assays, neonatal BN rat dermal cells were used as the necessary inducible cells in our modified skin reconstitution model (Fig. 4B). Distinguishing ours from other models, we selected natural SGS as the carrier and auxiliary scaffold. Before the graft, SGS was cut into circular patches by a C-shaped blade and sterilized (Fig. 4C). The mixture of GFP-labeled SG-captured keratinocytes, dermal cells, and Matrigel was prepared as a slurry and dropped onto the SGS patch (Fig. 4D). Owing to the properties of the SGS, we could simplify the grafting process and complete it in a few minutes without suturing (Fig. 4E). After 4 weeks, the new skin had formed in the grafted area, displaying dense and normal hair follicles (Fig. 4F). These results demonstrate that, on the one hand, our skin reconstitution assay is a simple and effective method for analyzing the skin-forming capacity of HFSCs and other epidermal lineage stem cells. On the other hand, these results revealed that SG-captured keratinocytes have the potential to reconstitute skin with the help of neonatal dermal cells.
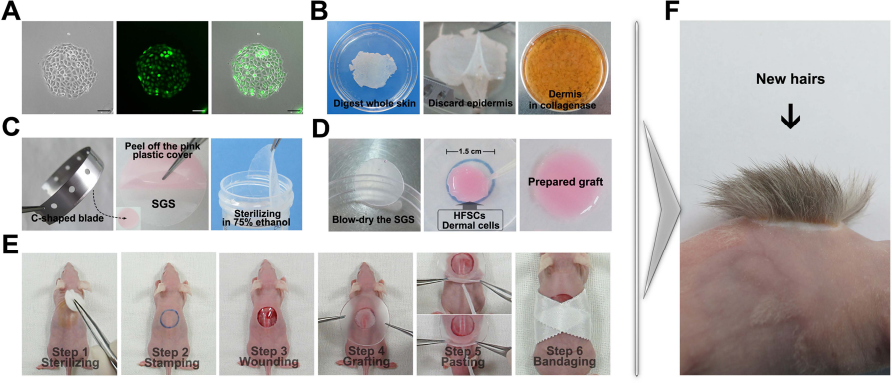
Skin reconstitution assay using SG-captured keratinocytes and neonatal rat dermal cells. (A) GFP-labeled SG-captured keratinocytes. (B) Preparation of neonatal rat dermal cells. (C) Preparation of SGS. (D) Preparation of the grafting mixture. (E) Grafting procedure. (F) Reconstituted skin growing on the wound area 4 weeks after graft. SGS, silicone gel sheeting; HFSCs, hair follicle stem cells. Scale bars: 100 μm.
Evaluation of the Reconstituted Skin
To confirm the contribution of SG-captured keratinocytes, we further examined the characteristics of the reconstituted skin. Under the excitation light, the wound area was distinguished from the surrounding tissue based on bright green fluorescence (Fig. 5A). We observed that the new hairs grew out from the GFP-positive area (Fig. 5B). In contrast, the control mice that were only grafted with neonatal dermal cells did not exhibit green fluorescence or new hair formation (Fig. 5C). To investigate the internal structures of the reconstituted skin, we performed tissue sectioning and PI staining. As indicated in Figure 5D, there were obvious GFP-positive structures in the transplantation site. The newly formed tissues expressing green fluorescence further confirmed the contribution of the SG-captured keratinocytes. The reconstituted skin contained intact epidermal structures, primarily the epidermis, hair follicles, and SGs, all of which showed strong green fluorescence (Fig. 5E). The skin surrounding the transplantation site showed no fluorescence signal, except for nonspecific fluorescence in the hair shafts (Fig. 5F). All of these results further verified the multipotency of SG-captured keratinocytes that were obtained from a limited sample of tail skin.

Histological evaluation of the reconstituted skin. (A) Green fluorescence of the graft area under excitation light. (B) Graft area reconstituted with SG-captured keratinocytes and neonatal dermal cells as viewed under a fluorescence stereoscope. The bright green fluorescence of the graft area (left). The bright field of the graft area (right) and a side view (lower right corner). (C) Observations of the graft area reconstituted only by neonatal dermal cells under the fluorescence stereoscope. The lack of green fluorescence in the graft area (left). The light field of the graft area (right) and a side view (lower right corner). (D) Lateral merged view of a part of the nude mouse back skin grafted with the SG-captured keratinocytes and neonatal dermal cells as stained by PI. (E) GFP-positive epidermis, hair follicles, and SGs in the reconstituted skin. (F) Surrounding control skin showing no green fluorescence except nonspecific fluorescence in the hair shaft. Epi, epidermis; HF, hair follicle; SG, sebaceous gland. Scale bars: 50 μm (D), 100 μm (E, F).
Discussion
In this study, we established a novel method for obtaining a subpopulation of HFSCs from limited samples of rat tail skin through the isolation of SGs. These SG-captured HFSCs expressed K14, p63, integrin α6, K15, and Plet1. Furthermore, in a modified skin reconstitution model, they are able to reconstruct new epidermis, hair follicles, and SGs in the back skin of nude mice. This study provides a new and high-output method for obtaining HFSCs without the consumption of a large amount of skin tissue and the need for intact or large hair follicles. Meanwhile, the skin reconstitution assay used in this study is an easy-to-use system for evaluating the capacity of HFSCs to reconstruct epidermal structures.
Typically, HFSCs have been isolated by FACS, which requires the use of fluorescent antibodies for cell surface markers9,38. However, for clinical purposes, FACS is not an ideal HFSC-harvesting method. First, the anoikis 39 and low survival rate caused by FACS can result in a low yield of pure HFSCs, which leads to a considerable length of time for harvesting enough cells for clinical use. Second, the source of skin biopsies suitable for FACS is limited in humans. To avoid these disadvantages, we aimed to establish a novel technology for obtaining abundant HFSCs from limited skin biopsy without the need for FACS. This technique allowed us to obtain a significant number of HFSCs from single vibrissae hair follicles29,30, avoiding the disadvantages of FACS. However, there are many other types of hair follicles that lack conspicuous and large architectures. In this study, we focused on the SGs, well-developed structures adjacent to the isthmus, that have already been deemed to be an HFSC pool in addition to the bulge7,20. The innovative method applied in our study, which captures one population of HFSCs by isolating adherent SGs, is more suitable for fragile and small hair follicles, such as those found in humans.
In the hair follicle ORS, stem cells have been named for where they reside, such as infundibulum stem cells 21 , upper isthmus stem cells22,23, and upper and lower bulge stem cells24,26. For instance, in the gap between the SGs and the bulge, there are at least the following two groups of HFSCs: stem cells in the upper isthmus and upper bulge stem cells. The upper residents, that is, the upper isthmus stem cells, are known to be Plet1 and Lrig1 positive22,23,40, while the lower group, that is, the upper bulge stem cells, specifically express Lgr6 24 . Many groups have revealed the locations and properties of these HFSC subpopulations in vivo; however, the obtaining and amplification of certain HFSC subpopulations in vitro have not yet been reported. Using isolated SGs as the capturer, we obtained one population of HFSCs residing in the upper isthmus. These SG-captured HFSCs express both general HFSC markers, such as K14, p63, and integrin α6, and the distinguishable upper isthmus HFSC markers Plet1 and Lrig1. Likewise, our long-term cultured SG-captured HFSCs differentiated into cells of the hair follicle, epidermis, and SGs in our skin reconstitution assay, exhibiting the typical multi potency of in vitro cultured HFSCs. However, the unambiguous confirmation of the upper isthmus as the origin of our SG-captured HFSCs requires other representative genes to distinguish upper isthmus stem cells in addition to the lineage tracing data, both of which are currently unavailable. Elucidating additional characteristics of these cells will necessitate further microarray research on the genomic and proteomic levels of SG-captured HFSCs.
Several hair follicle reconstitution models have been developed to verify the multipotency of epidermal stem cells, including HFSCs, such as the chamber 41 , flap 42 , patch 9 , splint 43 , and scaffold 44 models. Among these, the chamber model is widely used in hair follicle reconstitution; however, this method requires a complex operating process for grafting the HFSCs and dermal cells together into the wound in the back skin of an immunodeficient mouse. The long surgical time also increases the risk of infection in recipient animals. Subsequently, the up hat and down loop of the silicone chamber must be removed 1 week after surgery, and this is the time when the wound repair is occurring. Any second injury caused by removing the residual scaffold could also reduce the success rate of skin reconstitution. In addition, we have observed that when the chamber assay is used to transplant HFSCs, only hair follicle structures underneath the wound could be found 7 weeks after surgery 30 . It is obvious that the low success rate could mask some positive results. The recently described scaffold model provides a simplified method to perform skin reconstitution, and based on the existing grafting models, we developed a grafting method using flexible and waterproof SGS. Since the 1980s, SGS has had a wide range of clinical applications for hypertrophic scars and keloids 45 . In this model, SGS can easily encapsulate grafts in the wound site by its weak viscous side. Combined with medical glue, the suture process and the subsequent suture removal could be omitted. In our transplantation experiment (more than three repetitions with more than four mice for one experiment), there was more or less hair growth seen in every nude mouse back skin grafted with HFSCs. Therefore, the present SGS model makes the grafting operation much more simplified and efficient.
In conclusion, we obtained a population of HFSCs residing in the upper isthmus by isolating and organ culturing adherent SGs. These SG-captured HFSCs show the typical signature gene expression profiles of HFSC and can rebuild new epidermis, hair follicles, and SGs in our modified skin reconstitution assay. Given the well-developed SGs present in human scalp hair follicles, the present strategy could be useful for obtaining human HFSCs easily and safely and then used in combination with the mature surgical techniques such as follicular unit transplantation (FUT) and follicular unit extraction (FUE).
Footnotes
Acknowledgments
This work was supported by NSFC Grant 31471287 (to Enkui Duan) and the Strategic Priority Research Program of the CAS XDA04020202-20, XDA01010202, and XDA04020419 (to Enkui Duan). We are indebted to all of the members of Duan's group for their suggestions; to Yong Zhao at the Institute of Zoology, CAS, for assistance in the nude mice breeding; to Xinhua Lin at the Institute of Zoology, CAS, for advice on fluorescence stereoscopy; to Shiwen Li and Hua Qin at the Institute of Zoology, CAS, for access to the confocal microscope and advice; and to Tong Zhao at the Institute of Microbiology, CAS, for her support with FACS. The authors declare no conflicts of interest.