
Abstract
Human hair follicle cells, both bulge and dermal papilla cells, were isolated and cultured in a GMP cell factory, in order to obtain an in vitro hair follicle source for encapsulation end transplantation in alopecia regenerative cell therapy. An in vitro model, constituted by organotypic cultures of human skin sample, was set up to simulate the dermal–epidermal interaction between bulge cells and dermal papilla cells, evaluating the possible new follicles formation and the regenerative potentiality of these hair follicle cells. Both the bulge and dermal papilla cells show an excellent cellular proliferation as well as an abundant extracellular matrix production. The immunofluorescence investigation revealed the positivity of both cell lines to CK15 and CD200, whereas both cell lines were negative to CD71 and Oct-4. The pool of cultured bulge and dermal papilla cells was injected into the deep dermis; at day 28 of culture, some organized areas with a higher cell density can be observed: the cells self-organize into papilla-like lengthened aggregates. In samples in which the follicular cells have been seeded on the dermis surface, an epidermis-like homogeneous monolayer on the dermis surface can be seen, therefore showing a potentiality of these cells for epidermis regeneration. These data show the efficacy of a cellular isolation and amplification approach to obtain an in vitro human hair follicle regenerative source on industrial scale in a GMP cell factory. The results also proved an intrinsic potentiality of follicular cells to in vitro recreate the epidermis for tissue engineering purposes. Thus, it is feasible to produce bioengineered hair follicles in a GMP cell factory, for encapsulation and transplantation in alopecic patients.
Introduction
In our society hair has a great aesthetic and psychosocial importance and it is estimated that 88% of the male population is affected by alopecia or hair loss. Since the onset of these pathologies is usually early, from 18 to 20 years, people affected suffer significant psychological and social discomforts. There are many kinds of alopecia, whose pathogenesis is usually different and not univocal, but all these pathologies lead to a more or less severe hair loss, which is hard to remedy (9). At the moment there are pharmacological and surgical treatments to cure alopecia, but they all show limits of applicability and efficacy. The pharmacological treatments are useful only in the early phase of the diseases, but they show many side effects as a result of their active principles and their action mechanism, especially in long-term therapies. The surgical treatments are useful even in late phases of these pathologies, but their limits are, in addition to the invasive method, the size of donor area from which the hair follicles (HFs) used as auto-grafts in hair restoration are extracted, which are usually not large enough to cover the patient's alopecic regions, and the expense, which limits the number of patients who can use this treatment (1). Moreover, besides pathological alopecia, cicatricial alopecia caused by different noxae (trauma, congenital, lymphocytic, neutrophilic, infective pathologies) can occur. Unger et al. (15) propose guidelines in identifying when surgical excision or transplantation should be employed.
One answer to the limitations of treatments for alopecic pathologies could be provided by regenerative medicine, particularly through cell therapy, which aims to regenerate new, healthy, cycling hair follicles (13). Hair is a skin appendage with an ectodermic origin and may be considered as a particularly differentiated epithelium. There is no new hair follicle formation in the scalp after birth, but the importance of cellular epithelia–mesenchymal interaction has come to the fore by studying the embryonic hair development and hair physiological cycle. Both hair follicle embryonic generation and hair follicle physiological regeneration depend on such an interaction. A niche abundant in mesenchymal stem cells, called bulge region, is located near the sebaceous gland region of the follicle, during the so-called anagen phase, but it can be also found in other hair regions, depending on the hair development phase.
In fact, an ideal model has recently been proposed by Cotsarelis (2) and Tiede et al. (14): hair follicle is a stem cell niche, and niche-embedded stem cells can migrate during hair cycle as well as differentiate into different lineages. The bulge region is therefore one of several sites in which mesenchymal cells reside. After isolation and culture, bulge cells (BU) can in vitro regenerate new hair follicles, epidermis, and sebaceous glands. For this reason they may be useful for skin regeneration: after wounding, bulge cells migrate towards epidermis, participating in a short-term wound healing (2). Bulge cells, which have clear mesenchymal stem cell characteristics, can be advantageously employed in regenerative therapy (12), although the use of somatic stem cells can raise some questions about transdifferentiation (8).
Qiao et al. (10,11) verified that in vitro incubation of mixed follicular cells prepared from embryonic mice developed follicle-like structures called “proto-hairs”; after implantation, proto-hairs fully develop into normal hairs, that persist and grow indefinitely.
The three-dimensional culture of dermal papillae (DP) cells has recently been identified as a strategy to optimize alopecia cell therapy by transplantation (3–5,19); in particular, alginate is widely employed in tissue engineering to optimize cultures and mimic the in vivo microenvironment. Microencapsulated hair dermal papilla cells have been used as prototypes and applied in murine model with positive, promising results. Large-scale cell encapsulation technology has been recently validated also in a good manufacturing practice (GMP) compliant cell factory, and technological resources are available for large-scale cell encapsulation intended for advanced therapies (16).
The aim of this work is to identify the most suitable cell resources for follicular regeneration in human, in order to create an abundant source for hair regeneration. The human cell expansion for the bioengineered hair follicle production was performed in a GMP cell factory to obtain a safe, less expensive product for encapsulation and transplantation.
Materials and Methods
Cell Isolation and Culture in a GMP Cell Factory
Tissues and cell treatment procedures were performed in a “clean room” (class B, GMP guidelines). Each suite is supported by single-pass, positive pressure (60 Pa) HEPA filtered air, at a temperature of 18°C and relative humidity 50%. The flow suite is unidirectional, with entry and exit air locks. The clean room environment was continuously monitored by wireless probes in order to assure the same operating conditions during each process step, as required by the European Community Guidelines. Low contamination levels were guaranteed by differential pressures, absolute filtration systems, and unidirectional laminar air flow. Personnel were trained with respect to the GMP guidelines. All the steps were conducted in certified ISO Class 5 (Class 100) laminar flow biosafety cabinets. Environmental cleanliness controls were performed before operation in the unmanned state, and in the manned state during normal use. Microbiological control tests were performed “in operation,” using settle plates to control static air and contact plates with specific instrumentation to control dynamic air; surfaces and personnel (operator's gloved hands) were performed after critical operations.
The processed samples were pieces of scalp (about 40 × 15-mm strips) derived from the occipital or parietal region obtained from five informed male subjects. The use of human tissues was approved by the appropriate ethical committee. The age of the patients ranged from 34 to 75 years (mean age: 56.6 ± 10.6 years). After two/three rinses with Dulbecco's phosphate-buffered saline solution (PBS) with added antibiotics (gentamycin 50 μg/ml), the samples were trimmed in small pieces and incubated overnight at +4°C with 5 ml of Dispase II. From each sample, 50–100 follicles were recovered; hair follicles (HFs) were squeezed out carefully from the skin with sterile forceps, removing the excess adipose tissue below. Under the dissecting microscope, the HFs in anagen phase were selected and then sectioned lengthwise in three portions (Fig. 1). After an additional rinse with PBS with added antibiotics, the middle sections, containing the BU region, and the lower sections, containing the DP cells, were transferred into petri dishes (30–50 HFs or bulges/dish) for the primary culture, and immerged in hair follicle medium (HFM) composed of three parts of DMEM and one part of HAM's F12, supplemented with 10% of FBS, L-glutamine 0.584 mg/ml, gentamycin 50 μg/ml, sodium pyruvate 0.11 mg/ml, hydrocortisone 0.4 μg/ml, recombinant human insulin 5 μg/ml, EGF 10 ng/ml, and amphotericin B 2.5 μg/ml. The primary culture petri dishes were incubated at 37°C and 5% CO2, with medium change twice a week. After 3 weeks of primary culture both the BU cells and the DP cells were collected with 0.125% trypsin+ EDTA for 20 min at 37°C. The harvested cells were reseeded in tissue culture flasks for secondary culture (cell density: 2 × 104 cells/cm2) until confluence, which was reached in two weeks; after this period, cells were trypsinized as above and a tertiary culture was performed seeding cells at a density of 5–8 × 103 cells/cm2 until confluence. Morphological investigations were performed on both BU and DP cells after a parallel culture in chamber slides; immunofluorescent characterizations were performed on BU and DP cells at the confluence stage of the tertiary culture.

Hair sections. KR, keratinic region; BU, bulge region; DP, dermal papilla region. Original magnification: 5×.
Organotypical Culture Model
A full-thickness skin sample, from abdominoplasty surgery in informed patients, was used for the follicular and epidermis regeneration model. The skin sample was trimmed in small pieces of 3–5 mm and then incubated with Dispase II for the dermal–epidermal separation and placed on metal supports, for an organotypical culture (Fig. 2). Two confluence flasks from tertiary culture, one constituted of BU cells and one of DP cells, were trypsinized; counting and viability tests were performed for calculating cell densities of seeding. The BU cells were seeded at 5 × 105/cm2 and DP cells at 1 × 105/cm2. The cells were pooled together in a vial according to the proper densities; a small steel ring was placed on the pieces of skin to avoid the cell dispersion in the medium (Fig. 2), and the cell suspensions were slowly injected into the deep dermis or seeded on the surface. The skin samples were cultured for 28 days with HFM medium changed every 2 days. After culture, histological investigations were performed.

Organotypical culture.
Histological Investigation
Chamber slide-cultured cells were fixed in 35% formaldehyde and stained with hematoxylin-eosin; organotypical-cultured tissues were fixed in 35% formaldehyde, dehydrated using alcohol scale, embedded in paraffin, cut into 5-μm-thick sections, and treated with hematoxylin-eosin stain or trichromic Masson stain.
Immunofluorescence Staining
Immunofluorescence investigation was performed by laser confocal microscopy. Samples were fixed in 35% formaldehyde in PBS solution, dehydrated by alcohol scale, and mounted on immunofluorescence microscope slides. The sections were treated with a 1% Triton X-100 solution in PBS medium; in order to block aspecific binding sites, samples were treated with 2% bovine serum albumin in PBS medium and left to stand for 30 min at room temperature. After this period, samples were treated (37°C for 60 min in light proof conditions) with primary antibody against CD200, CK15, CD71, and Oct-4. Primary antibodies were diluted in PBS medium with bovine serum albumin 1%. The samples were further treated with the secondary dye-coupled antibody, Goat anti-mouse IgG-Rhodamine (TRITC) conjugate (1:200 dilution, Chemicon). The slides were then treated with Vectashield liquid mounting media and DAPI. Slides were observed under a Leica confocal fluorescence microscope with oil immersion, 63× objective, 1.32 NA aperture. For each field, three confocal planes were acquired, and the elaboration of planes was performed by FluoView (Leica) software. Images were stored as single TIFF 24-bit with three distinct channels for red, green, and blue.
Results
The results for the microbiological environmental tests before and during tissue and cell processing were within the limits fixed by GMP guidelines for contamination in advanced therapy products.
Both the BU and DP cells show an excellent cellular proliferation as well as an abundant extracellular matrix production (Figs. 3 and 4); the DP cells show an organization characterized by ring structures, with an empty zone inside (Fig. 4).
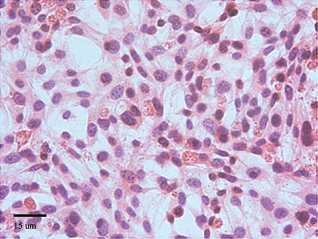
Bulge cells: secondary culture, 7 days, hematoxylin/eosin stain. Original magnification: 25×.

Microphotograph of dermal papilla cells: secondary culture, 14 days, hematoxylin/eosin stain. Original magnification: 10×.
The immunofluorescence investigation revealed, after the third confluence step, the positivity of both BU and DP cells to CK15 and CD200, whereas both cell lines were negative to CD71 and Oct-4. Positivity to CK15 and CD200 in DP cells is shown in Figure 5.
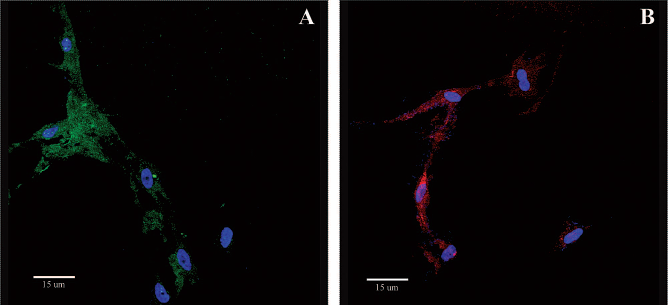
Fluorescent microphotographs of dermal papilla cells, at the confluence stage of the tertiary culture. DAPI (blue). Original magnification: 63×. (A) CD200+/FITC (green); (B) CK15+/TRITC (red).
The pool of cultured BU and DP cells was injected into the deep dermis; at day 28 of culture, some organized areas with a higher cell density can be observed: the cells self organize into lengthened aggregates (Fig. 6A). In samples in which the follicular cells have been seeded on the dermis surface, a homogeneous monolayer on the dermis surface can be seen, showing a potentiality of these cells for tissue regeneration (Fig. 6B).

(A) Follicular cells injected into the deep dermis, 28 days, trichromic Masson stain. Original magnification: 10×. (B) Follicular cells seeded on the surface, 28 days, trichromic Masson stain. Original magnification: 25×.
Discussion
Hair follicle has been described as an actual invaginated “miniature organ” with a number of cell lineages, including mesenchymal stem cells, and paracrine signaling molecules (17). Bulge cells are situated in a region near the nerve, muscle, sebaceous, and melanocyte components of skin but can be found also in the matrix region, located at the bottom of the HF: if BU cells migrate along the follicle during the anagen phase of hair cycle, or supply the matrix cells by renewing, is matter of debate (2). The experimental evidence indicates that both BU and DP cells cultured and expanded in vitro present stem cell characteristics in terms of continuous renewal and high proliferative capacity: they reach the confluence in a brief time and at the third cycle stem cell markers are preserved, as can be seen by the presence of CD200 and CK15. The positivity of CK15 and CD200 are paramount to identify hair follicle stem cells, as reported by Lyle et al. (6) and Ohyama et al. (7), for K15 and CD200, respectively. Moreover, negativity to CD71 could reflect the bulge origin of cultured cells, as mentioned by Ohyama et al. (7); negativity to OCT4 could be an index of the lack of “embryonic” SC transcription factors, as reported by Yu et al. (18).
This is, to our knowledge, the first study proving the feasibility of a safe and efficacious methodology that could contribute for future important clinical applications on alopecia disease. These data show the efficacy of a cellular isolation and amplification approach to obtain an in vitro human hair follicle regenerative source on industrial scale in a GMP cell factory. These cells can be used in skin organotypic culture as cell implantation prototype for follicular regeneration. The results also proved an intrinsic potentiality of follicular cells to in vitro recreate the epidermis for tissue engineering purposes. Thus, it is feasible to produce bioengineered hair follicles in a GMP cell factory, for encapsulation and transplantation in alopecic patients. Further studies could be designed in order to better understand the capacity of different skin compartments in regenerate new hair follicles.
Footnotes
Acknowledgments
This work was supported by Pavia and Milan Universities and by Regime Scozzese Rettificato-Giurisdizione Italiana. The authors thank M.D. Vincenzo Gambino (Istituto Medico Quadronno, Milan, Italy) and D. Patrizia Vaghi (Centro Grandi Strumenti, Pavia University).