
Abstract
Neural stem cells (NSCs) promote recovery from brain trauma, but neuronal replacement is unlikely the sole underlying mechanism. We hypothesize that grafted NSCs enhance neural repair at least partially through modulating the host immune response after traumatic brain injury (TBI). C57BL/6 mice were intracerebrally injected with primed human NSCs (hNSCs) or vehicle 24 h after a severe controlled cortical impact injury. Six days after transplantation, brain tissues were collected for Western blot and immunohistochemical analyses. Observations included indicators of microglia/macrophage activation, M1 and M2 phenotypes, axonal injury detected by amyloid precursor protein (APP), lesion size, and the fate of grafted hNSCs. Animals receiving hNSC transplantation did not show significant decreases of brain lesion volumes compared to transplantation procedures with vehicle alone, but did show significantly reduced injury-dependent accumulation of APP. Furthermore, intracerebral transplantation of hNSCs reduced microglial activation as shown by a diminished intensity of Iba1 immunostaining and a transition of microglia/macrophages toward the M2 anti-inflammatory phenotype. The latter was represented by an increase in the brain M2/M1 ratio and increases of M2 microglial proteins. These phenotypic switches were accompanied by the increased expression of anti-inflammatory interleukin-4 receptor α and decreased proinflammatory interferon-γ receptor β. Finally, grafted hNSCs mainly differentiated into neurons and were phagocytized by either M1 or M2 microglia/macrophages. Thus, intracerebral transplantation of primed hNSCs efficiently leads host microglia/macrophages toward an anti-inflammatory phenotype that presumably contributes to stem cell-mediated neuroprotective effects after severe TBI in mice.
Keywords
Introduction
Traumatic brain injury (TBI) is a major cause of mortality and morbidity, especially among young adults, and lifelong disability is common in those who survive. Despite considerable progress, clinical treatment is still limited to supportive care. Stem cell therapy has received much attention as a potential means of promoting recovery after various injuries to the central nervous system (CNS). For TBI in particular, therapy using embryonic, fetal, or adult stem cells has shown beneficial effects in animal models1–6, with some being used in clinical trials 7 . Although stem cell-mediated neural cell replacement may be one beneficial effect, other mechanisms such as providing trophic support and suppressing inflammation are also attributed to the beneficial effects of stem cells in mediating systemic homeostasis and facilitating neural repair8,9. Thus, an understanding of the mechanisms underlying stem cell-mediated benefits is important for future development of novel and more effective treatments for TBI.
Anti-inflammation measures have been increasingly appreciated as one of the important mechanisms for the beneficial effects of cell transplantation. Neuroinflammation, characterized by activation of microglia/macrophages and astrocytes as well as production of various cytokines, occurs acutely after traumatic injury and is one major factor contributing to the secondary injury in the CNS10–12. Microglia, the macrophages in the brain and spinal cord representing 5% to 10% of the total glial population, are the resident innate immune cells in the CNS and are activated by trauma 13 . Peripheral macrophages can also infiltrate into traumatically injured areas. Injury-induced activation of microglia/macrophages has a dual role that either exacerbates secondary injury or promotes neural repair14–19. These divergent effects may be attributed to the distinct microglial subsets: “classically activated” proinflammatory (M1) or “alternatively activated” anti-inflammatory (M2) cells. M1 microglia/macrophages and their associated cytokines are neurotoxic, whereas M2 promotes a regenerative response in injured CNS 16 .
TBI activates both classic M1 and alternative M2 phenotypes. However, M2 activity decreases within a few days after injury, whereas M1 remains high for much longer and intensifies damage following TBI over time18,20. Thus, promoting the M2-mediated anti-inflammatory response while reducing M1-associated proinflammatory effects should be a good strategy to improve repair14,15. In this regard, we showed that systemic delivery (intravenous) of human bone marrow-derived multipotent adult progenitor cells (hMAPCs) induced an increase of the M2/M1 ratio in mouse brains after controlled cortical impact (CCI) injury 21 and suggested this contributed to cell-mediated neuroprotection 22 . We have also observed improved cognitive function after transplanting another type of stem cell, human neural stem cells (hNSCs), locally into injured rodent brains23,24. Immunomodulation has been suggested as one mechanism of these NSC-mediated beneficial effects 25 . However, it is unknown whether locally grafted hNSCs modulate host microglia/macrophages in a way similar to those systemically injected hMAPCs. The aim of the present study is to determine how intraparenchymally grafted hNSCs affect resident microglia and macrophage infiltration, and whether this focal cell transplantation ameliorates neuropathological changes in a mouse model of TBI. To minimize interference from immunosuppressive treatments and to make a comparison with our previous hMAPC transplantation studies, we did not use immunosuppressants in this human to mouse xenotransplantation study.
Materials and Methods
Animals and TBI
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas Health Science Center. A total of 33 adult male C57BL/6 mice (6–7 weeks of age; Harlan Laboratories Inc., Houston, TX, USA) were used for 7-day survival assessments after CCI injury. The animals were randomly divided into three groups: sham injury (craniectomy without injury, n = 11), CCI injury plus vehicle injection (CCI + Veh, n = 11), and CCI injury plus primed hNSC grafting (CCI + hNSCs, n = 11). An additional three mice were included in an initial experiment and sacrificed at 1 day postinjury. A CCI device [eCCI Model 6.3; Virginia Commonwealth University (VCU), Richmond, VA, USA] was used to administer a unilateral brain injury as described previously 26 . Male mice weighing 17 to 21 g were anesthetized with 4% isoflurane [University of Texas Medical Branch (UTMB) Clinical Chemistry, Galveston, TX, USA]/O2, and the head was mounted in a stereotactic frame. Animals received a single impact of 1.0-mm depth of deformation with an impact velocity of 5.0 m/s and a dwell time of 150 ms (severe injury), making the impact to the right parietal association cortex (midway between bregma and lambda, 1 mm right to the midline). Sham injuries were performed by anesthetizing the animals, making a midline incision, and separating the skin, connective tissue, and aponeurosis from the cranium. The incision was then closed 27 .
Preparation and Transplantation of Primed hNSCs
The primary K048 line of hNSCs was a gift from Dr. Clive N. Svendsen 28 , which was originally derived from the forebrain of an 8-week human fetus following procedures approved by the Local Research Ethics Committee of the University College Hospital (London, UK), and the recommendation by the Polkinghorne Committee and the guidelines of the Department of Health in the UK.
The cells were maintained without genetic modifications for up to 112 passages in vitro as described in our previous publication with minor modifications 29 . Briefly, K048 cells were cultured in basic medium (see below) supplemented with 20 ng/ml epidermal growth factor (EGF; R&D Systems Inc., Minneapolis, MN, USA), 20 ng/ml basic fibroblast growth factor (bFGF; R&D Systems), 5 μg/ml heparin (Invitrogen, Life Technologies, Carlsbad, CA, USA), 10 ng/ml leukemia inhibitory factor (LIF; Chemicon International Inc./Fisher Scientific, Pittsburgh, PA, USA), and N2 (100 μg/ml transferrin, 100 μM putrescine, 20 nM progesterone, 30 nM sodium selenite; all from Sigma-Aldrich, St. Louis, MO, USA). The basic medium was composed of Dulbecco's modified Eagle's medium (DMEM; CellGro; Mediatech Inc., Manassas, VA, USA)/F12 (Invitrogen) (3:1), 15 mM HEPES (Sigma-Aldrich), 1.5% glucose (Sigma-Aldrich), 2 mM L-glutamine (Sigma-Aldrich), and penicillin/streptomycin (Sigma-Aldrich). Cells were incubated with 8.5% CO2 at 37°C. Two thirds of the growth medium was replaced every 3 to 4 days. Expanded neurospheres were dissociated into single cells once every 7 days with 0.025% trypsin (Sigma-Aldrich) and mechanical trituration. For transplantation of hNSCs, neurospheres (3 days after passage, passages 20–30 or 200–300 days in vitro) were seeded at 6 × 104 cells/cm2 in a T25 culture flask precoated with 0.01% poly-D-lysine (Sigma-Aldrich) and 0.5 μg/cm2 mouse laminin (Invitrogen). Cells were primed with 10 ng/ml bFGF, 2.5 μg/ml heparin, and 1 μg/ml laminin (FHL) for 5 days and then switched to DMEM/F12 media containing B27 (Invitrogen) for 1 day. Prior to transplantation, primed hNSCs were dissociated by incubation with 0.025% trypsin/200 U/ml DNase (Sigma-Aldrich) for 10 min at 37°C followed by mechanical trituration. Numbers of live cells were determined by a trypan blue (Sigma-Aldrich) exclusion assay. Cells were centrifuged at 1,100 × g, resuspended in basic medium supplemented with B27, 250 U/ml DNase, and 0.6% glucose at a density of 0.5 × 105 cells/μl, and then stored on ice until transplantation. To track grafted hNSCs, some cells were prelabeled with recombinant adeno-associated viral (AAV) vectors containing an enhanced green fluorescent protein (AAVegfp; the Vector Core at the University of North Carolina, Chapel Hill, NC, USA) 5 days before transplantation. The labeling efficiency was around 80%. Cells were prepared and transferred from Dr. Wu's laboratory at UTMB to Dr. Grill's laboratory at the University of Texas (UT) Health Science Center on the day of grafting.
One day after CCI injury, mice were transferred from the Cox laboratory at UT-Health to Dr. Grill's surgery room (also at UT-Health). CCI mice were anesthetized using a combination of ketamine and xylazine (both from Henry Schein Animal Health, Fort Worth, TX, USA) 30 . Fifty thousand primed hNSCs or vehicle alone (2 μl medium used to resuspend hNSCs; see above) were slowly injected over a 3-min period of time rostral and slightly medial to the injury site in the cortex using a 5-μl Hamilton syringe with a blunt 27-gauge needle held in place in a stereotactic frame (David Kopf Instruments, Tujunaga, CA, USA). The needle was held in place for an additional 3 min after injection before gradually withdrawing.
Histological Analyses
Five animals in each group were perfused with 4% paraformaldehyde (PFA; Sigma-Aldrich) 6 days after grafting. Mouse brains were postfixed overnight and then immersed in 30% sucrose until they sank. Tissues were embedded in OCT compound (Thermo Fisher Scientific, Waltham, MA, USA). Serial transverse sections at a thickness of 30 μm were cut with a cryostat (Leica CM1900; Meyer Instruments Inc., Houston, TX, USA). Every fifth section throughout the rostral–caudal axis of the brain was stained with hematoxylin and eosin (H&E; Sigma-Aldrich) to determine overall structural damage, or with various antibodies to characterize microglia and grafted cells. Sections were incubated for 1 h in 5% normal serum plus 0.3% bovine serum albumin (BSA; Sigma-Aldrich) and 0.25% Triton X-100 (Sigma-Aldrich) in Tris-buffered saline (TBS; Sigma-Aldrich). This blockage of nonspecific binding and permeabilization procedure was followed by an overnight incubation with primary antibodies at optimal concentrations at 4°C. After three 10-min rinses with TBS, samples were incubated with species-specific secondary antibodies conjugated with either Alexa Fluor 568 (red) or Alexa Fluor 488 (green) fluorophores (1:500 dilution; Molecular Probes, Eugene, OR, USA) for 3 h at room temperature (RT) in the dark. After three 10-min rinses with TBS, sections were counterstained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 5 min at RT and then mounted with Fluoromount G (Thermo Fisher Scientific). Primary antibodies included Iba1 (#019-19741; Thermo Fisher Scientific) at 1 μg/ml, CD86 (BD Biosciences, San Jose, CA, USA) at 2.5 μg/ml, CD16/32 (BD Biosciences) at 0.2 μg/ml, arginase 1 (V-20) (Santa Cruz Biotechnology Inc., Dallas, TX, USA) at 1 μg/ml, CD206 (R&D Systems) at 1 μg/ml, green fluorescent protein (GFP; Aves Labs Inc., Tigard, OR, USA) at 1:500, glial fibrillary acidic protein (GFAP; EMD Millipore, Billerica, MA, USA) at 1:2,000, and doublecortin (DCX; Abcam, Cambridge, MA, USA) at 1 μg/ml. To detect axonal damage, tissues were labeled with an antibody against the amyloid precursor protein (APP; Invitrogen) at 1.25 μg/ml. All images were taken using Nikon D-Eclipse C1si Laser Scanning Confocal system (Nikon, Tokyo, Japan).
Western Blot Analyses
Fresh half-brain tissues (ipsilateral to the injury) were collected from six mice per group to quantitatively determine the changes of various proteins on the injured side of the brain following treatments. Western blot analyses were performed as previously described 24 . For tissue collection, anesthetized mice were perfused with 5 ml of phosphate-buffered saline (PBS; Sigma-Aldrich) containing 1 mM phenylmethylsulfonyl fluoride (PMSF; Alexis Biochemicals, Farmingdale, NY, USA), 1× protease inhibitor cocktail (Sigma-Aldrich), 30 mM sodium fluoride (Sigma-Aldrich), and 1 mM sodium vanadate (Sigma-Aldrich). Immediately following decapitation, brains were removed and frozen in liquid nitrogen. Tissues were then homogenized in lysis buffer containing 25 mM HEPES (Sigma-Aldrich), 0.1% Triton X-100 (Thermo Fisher Scientific), 10% glycerol (Sigma-Aldrich), 1% protease inhibitor cocktail (Sigma-Aldrich), 30 mM sodium flouride (Sigma-Aldrich), and 1 mM sodium vanadate (Sigma-Aldrich), and centrifuged at 20,000 × g at 4°C for 20 min. Protein concentrations were determined using the Bio-Rad Protein Assay Dye (Bio-Rad Laboratories, Hercules, CA, USA). Total protein extracts were electrophoresed through NuPAGE 4–12% Bis-Tris gels (Invitrogen) and transferred onto nitrocellulose membranes (Bio-Rad). Following blocking, membranes were probed with primary antibodies at 4°C overnight and then with peroxidase-conjugated secondary antibodies (1:2,000; GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). Primary antibodies included APP (1:500; Invitrogen); CD86, a marker for M1 microglia (BD Biosciences) at 2.5 μg/ml; CD16/32 (BD Biosciences) at 0.2 μg/ml; arginase 1 (Santa Cruz Biotechnology) at 1 μg/ml; CD206 (R&D Systems) at 1 μg/ml; interleukin-4 (IL-4) receptor α (IL4Rα) (R&D Systems) at 0.1 μg/ml; and interferon-γ (IFN-γ) receptor β (IFNγRβ) (Abcam) at 1 μg/ml. Blots were reprobed with internal control monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1,000; Santa Cruz Biotechnology). Chemiluminescent detection of immunoreactive signals was assisted by ECL Hyperfilm (Amersham Biosciences Corp., Piscataway, NJ, USA) and subjected to densitometry analyses using AlphaEaseFC software (Alpha Innotech Co., San Leandro, CA, USA).
Quantitative and Statistical Analyses
Lesion volumes were determined according to the previously described method 31 . Briefly, every fifth section through the injury was mounted onto glass slides in sequential order, stained with hematoxylin (Vector Laboratories, Burlingame, CA, USA), and coverslipped with an aqueous medium (Fluoromount G). Slides were subsequently placed on a flatbed scanner (LaserJet Pro CM1415fnw; HP, Miami, FL, USA), scanned at a resolution of 1,200 dots per inch (DPI), and saved as tagged image file format (TIFF) files. Five mice per group were included for quantitative analyses using ImageJ software [National Institutes of Health (NIH), Bethesda, MD, USA]. Lesion area per section was generated by manually outlining each lesion cavity. Lesion volume was then determined as the sum of the area measurement of each section × the thickness of the section (30 μm) × the sampling interval. These assessments were performed by an investigator blinded as to treatment.
For confocal imaging of brain sections, three to four mice per group per marker were used for quantitative immunohistochemical analyses. Every fifth section for a total of three sections per mouse, with four mice per group, were used for quantitation of the intensity of Iba1 immunofluorescent staining around the injury site and blindly analyzed. The region of interest (ROI) was manually defined at the edge of area with positive Iba1 staining. The relative intensity of Iba1 labeling, expressed as the percentage of pixels in the ROI above the threshold per section, was automatically analyzed by ImageJ software. Numbers of cells stained with various microglial markers in the sample region (i.e., around the lesion site, ~2 mm in diameter in the ipsilateral cortex) were determined stereologically using a fractionator analysis method 32 . Briefly, six to seven sections (30 μm, every 20th section) per mouse brain (a total of 130–140 serial sections) were taken in a uniform random pattern. Upper and lower boundaries of optical dissectors (grid size of 100 × 100 μm, height of 30 μm) were set at appropriate confocal planes, with attention to three-dimensional exclusion and inclusion lines. The total number of a specific marker-labeled cells was averaged among three mice each group. APP+ profiles (>5 μm) were also counted stereologically and averaged from four mice per group.
For statistical analyses, data were expressed as means ± SEM and analyzed by Student's t-test or one-way analysis of variance (ANOVA) with Tukey's or Newman–Keul post hoc tests. Data were analyzed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA). Results were considered significant at p < 0.05.
Results
Transplantation of Primed hNSCs Reduces Abnormal APP Accumulation Following TBI
Primed hNSCs were used in our transplantations because these cells tended toward the neuronal progenitor lineage and have shown neuroprotective effects in our previous transplantation studies in rats with moderate fluid percussion brain injury23,24. To determine direct effects of primed hNSC grafts on modulating innate immune responses following TBI, we did not use any immunosuppression protocols. Brains tissues were collected 7 days after severe CCI injury (6 days posttransplantation) and subjected to H&E staining and immunostaining. The time point of 7 days postinjury was chosen based on the previous literature demonstrating that the ratio of inflammatory (M1) over anti-inflammatory (M2) phenotypes peaked around 7 days postinjury 16 . Our question was whether hNSC transplantation reverses the M1/M2 ratio at this peak time point.
Because of large variations in severe CCI, we could not detect significant changes of median lesion volumes between the vehicle control and hNSC-transplanted mice. However, we did observe an accumulation of APP immunoreactive profiles (>5 μm in size) in the injured mouse brain, indicating considerable axonal injury, when compared to the negative APP staining in sham mice (Fig. 1A). This abnormal APP accumulation was significantly decreased by 50% after transplantation of primed hNSCs (Fig. 1B–D). Western blot analyses also revealed a TBI-induced 40% APP increase in vehicle controls, which was reversed by cell transplantation (Fig. 1E). Further more, cell transplantation reduced post-TBI increases in expression of α smooth muscle actin (α-SMA) (Fig. 1F), an indicator of abnormal cytoskeletal accumulation detected only after injury in neurons as we reported previously 24 .

Transplantation of primed human neural stem cells (hNSCs) reduces axonal injury in mice after TBI. (A–D) Immunofluorescent staining of mouse brain sections collected 7 days after controlled cortical impact (CCI) injury. No specific amyloid precursor protein (APP) accumulation was observed in the brains of sham animals (A). Representative images of APP in regions near the injury site are shown from traumatic brain injury (TBI) + vehicle (Veh) (B) and TBI + Cell (C). Scale bar: 10 μm. (D) Stereological analyses show that hNSC transplantation significantly reduces the number of APP-labeled profiles (>5 μm, arrows in B and C). *p < 0.05, n = 5, t-test. (E–F) Western blot analyses of protein extracts from the ipsilateral mouse brains 7 days postinjury. TBI significantly increased the expression of APP and α smooth muscle actin (α-SMA), which were both reduced by cell transplantation. GAPDH, glyceraldehyde-3-phosphate dehydrogenase, internal control. Values are means ± SEM, n = 6. #p < 0.05 compared to sham; **p < 0.01 and *p < 0.05 compared to T + Veh (CCI-induced TBI + vehicle); CCI-induced TBI plus primed hNSC grafting (T + Cell) one-way analysis of variance (ANOVA) with Tukey's or Newman–Keul tests.
Transplantation of Primed hNSCs Attenuates TBI-Induced Microglial/Macrophage Activation in Mouse Brain
To determine the immune response to cell transplantation after TBI, we focused on microglia/macrophages in the mouse brain 7 days postinjury. Iba1 was used as a marker to detect microglia/macrophages in mouse brains. In sham animals, Iba1-labeled microglia were scattered throughout the brain, were typically small cells, and had ramified morphologies (Fig. 2A and B). Seven days after TBI, intensive labeling of Iba1 accumulated around the injury site (Fig. 2C), most of which exhibited an amoeboid morphology (Fig. 2D). To further explore the effect of hNSC transplantation on microglial/macrophage proliferation and activation, we selected serial brain sections from the hNSC- and vehicle-injected groups, and then measured the intensity of Iba1 staining area using the NIS-Elements BR software. Compared to the vehicle controls, hNSC transplantation significantly reduced the intensity of Iba1 immunoreactivity by 14% (Fig. 2E–G). By costaining with markers for microglial subtype M1 (CD16/32 and CD86) and M2 (arginase 1 and CD206), we found that these Iba1+ cells were double labeled with one of those antibodies in both the vehicle (Fig. 2H–K) and the treatment (data not shown) groups. No differences in terms of the morphology of microglia were detected between the vehicle and cell transplantation groups (see also below).

TBI and hNSC transplantation oppositely affect the activation of microglia/macrophages in mouse brains. Immunostaining with Iba1 shows the morphology of resting microglia in the sham brain (A, B), and amoeboid-like microglia/macrophages accumulated surrounding the injury site at 7 days after CCI injury (TBI + Veh) (C, D). The arrows in (A) and (C) are enlarged in (B) and (D), respectively. Compared with vehicle controls (E), transplantation with primed hNSCs significantly reduced the intensity of Iba1-labeled microglia (F, G), n = 3–4, mean ± SEM, **p < 0.01, t-test. (H–K) Confocal images of Iba1+ microglia colabeled with M1 markers, CD16/32 (H) and CD86 (I), or with M2 markers, arginase 1 (J) and CD206 (K). Scale bars: 500 μm (A, C, and E–F), 10 μm (B, D, and H–K).
Primed NSC Transplantation Promotes a Transition From M1-Dominant to M2-Dominant Microglial/Macrophage Polarization in the Injured Mouse Brains
To determine the effect of hNSCs on microglial polarization after TBI, we carried out both Western blot and immunofluorescent analyses to assess changes in the M1 and M2 microglial phenotypes. Antibodies included CD86 and CD16/32 for M1, as well as CD206 and arginase 1 for M2. Overall expressions of these marker proteins were quantified by Western blotting on the ipsilateral brain samples from six mice per group. Because the majority of microglia/macrophages were located near the site of injury, histological analyses were performed by stereologically counting the cells labeled with M1 or M2 markers surrounding the injury site in three mice per group.
For M1 markers, Western blotting showed that TBI resulted in a significant increase of CD16/32 and a slight increase of CD86 in the ipsilateral mouse brain at 7 days postinjury, whereas hNSC transplantation significantly reduced both markers (Fig. 3A and G). Immunohistochemistry did not detect an intense labeling of CD16/32 or CD86 in the brains of sham-operated animals. However, in animals after TBI, CD16/32- and CD86-labeled large round cells accumulated around the injury site (Fig. 3C–F and I–L). Stereological analyses indicated that the numbers of M1 marker-labeled cells tended to decrease 6 days after hNSC grafting (or 7 days postinjury) (Fig. 3B–F and H–L).

Transplantation of hNSCs reduces M1 phenotypic activation in mouse brains after TBI. (A) Western blot analyses show significant increases of CD16/32 expression in the injured hemisphere (T + V), which is reduced by hNSC transplantation (T + C). (B) Stereological analyses suggest that cell transplantation tends to decrease the total number of CD16/32-immunostained cells that surround the injury site. Representative fluorescent images of brain tissues immunostained with a CD16/32 antibody in vehicle control (C) and cell transplantation (D), which are enlarged in (E) and (F), respectively. (G) Western blot analyses do not show a significant increase of CD86 expression in the injured hemisphere, but cell transplantation reduces the level of CD86 when compared to TBI controls. (H) Stereological analyses suggest that cell transplantation tends to decrease the total number of CD86-immunostained cells surrounding the injury site. Representative fluorescent images of brain tissues immunostained with a CD16/32 antibody in vehicle control (I) and cell transplantation (J), which are enlarged in (K) and (L), respectively. (A, G) Values are expressed as means ± SEM, n = 6, *p < 0.05, t-test or one-way ANOVA with Tukey's test. (B, H) Mean ± SEM, seven sections/mouse, three mice/group, p = 0.17 in (B) and p = 0.14 in (H) when compared to T + V, t-test. Scale bars: 100 μm (C, D, I, and J), 20 μm (E, F, K, and L).
For M2 markers, sustained increases in the levels of CD206 and arginase 1 were evident in the ipsilateral brains of TBI mice and particularly in those mice with TBI plus hNSC transplantation (Fig. 4A and H). Immunofluorescent staining detected CD206 mainly in the meninges of sham brains (Fig. 4C) and occasionally in the parenchyma (arrowhead in Fig. 4C). At 7 days post-TBI, intense CD206+ cells were detected around the injury site in the parenchyma and perivascularly (arrowhead in Fig. 4D and F). Arginase 1-labeled cells were not detectable in sham-operated brains but were detectable after injury (Fig. 4J and L). Transplantation of hNSCs further increased the numbers of CD206 and arginase 1 immunoreactive cells (Fig. 4B, E, G, I, K, and M).

Transplantation of hNSCs increases M2 phenotypic expression in mouse brains after TBI. (A) Western blot analyses show that TBI does not significantly increase CD206 expression in the injured hemisphere (T + V), but after hNSC transplantation there is a significant increase 7 days postinjury (T + C). (B) Stereological analyses show a trend increase of the total number of CD206-labeled cells after TBI, which is further increased by cell transplantation. Representative fluorescent images of brain tissues immunostained with a CD206 antibody in sham (C), vehicle control (D), and cell transplantation (E); the latter are enlarged in (F) and (G), respectively. Arrowheads, CD206+ cells along vessels. Note that CD206+ cells are detected mainly in the meninges or perivascularly in the intact brain (C). (H) Western blot analyses do not detect significant increases of arginase 1 protein expression in the injured hemisphere after TBI or hNSC transplantation. (I) Stereological analyses suggest that cell transplantation tends to increase the total number of arginase 1-immunostained cells that surround the injury site. Representative images of brain tissues immunostained with an arginase 1 antibody are shown in vehicle control (J) and cell transplantation (K), which are enlarged in (L) and (M), respectively. (A, H) Mean ± SEM, n = 6, **p < 0.01, one-way ANOVA with Tukey's test. (B, I) Mean ± SEM, seven sections/mouse, three mice/group, *p < 0.05 and p = 0.12 when compared to T + V, respectively; one-way ANOVA or t-test. Scale bars: 100 μm (C–E, J, and K), 20 μm (F, G, L, and M).
To further clarify the effect of hNSC transplantation on the phenotypic polarization of microglia/macrophages after TBI, we calculated and compared the percentages of M1 and M2 subsets for each mouse. As shown in Figure 5A, a much higher percentage of M2 cells is evident in the transplantation group than in the vehicle control, 72.1% versus 42.3%. More specifically, transplantation of hNSCs resulted in a 48.4% reduction of M1 cells and an 81.8% enhancement of M2 (Fig. 5B and C), indicating that hNSC transplantation promoted a phenotypic transition toward the M2 phenotype.

Transplantation of hNSCs promotes a transition of microglia/macrophages from M1 to M2. (A) Ratios of M2 (arginase 1 plus CD206) and M1 (CD16/32 plus CD86) over the total number of M1 and M2 cells (M2/M1 + M2 and M1/M1 + M2) in the injury site 7 days post-TBI. Stereologically counted total numbers of M1 (B) and M2 cells (C) in TBI plus vehicle injection (T + V) versus TBI plus hNSC transplantation (T + C). Values are expressed as means ± SEM, n = 3. *p < 0.05, **p < 0.01, t-test.
Classically activated M1 microglia/macrophages are induced by IFN-g alone or in concert with microglial stimuli or cytokines, whereas alternative M2 activation is induced by interleukins such as IL-4. As an initial step to elucidate the underlying mechanisms of the M1–M2 transition, we focused on the receptors for proinflammatory IFN-γ (IFN-γRβ) and anti-inflammatory IL-4 (IL-4Rα). Western blot analyses showed a trend of IFN-γRβ increase in mice with TBI plus vehicle injection, which was significantly reduced by hNSC transplantation (Fig. 6A). On the other hand, even though TBI increased the level of IL-4Rα, cell grafting further augmented its expression in the injured brain (Fig. 6B). In an in vitro setting of coculturing hNSCs with the microglia isolated from rodent brains, we found that hNSCs turned microglia from amoeboid to ramified morphologies, indicating a direct effect of hNSCs on microglial phenotypes (data not shown).
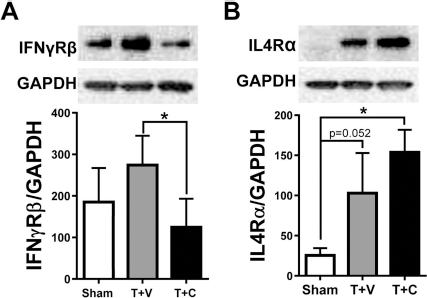
Transplantation of hNSCs modulates receptors for both proinflammatory and anti-inflammatory cytokines in mouse brains. Western blot analyses were performed on proteins extracted from mouse brains at 7 days post-injury. TBI with vehicle injection tends to increase the levels of both proinflammatory interferon-γ receptor β (IFN-γRβ) (A) and anti-inflammatory interleukin-4 receptor α (IL-4Rα) (B). Cell transplantation reduces IFN-γRβ but further increases IL-4Rα. Values are expressed as means ± SEM, n = 6, *p < 0.05, one-way ANOVA with Tukey's tests.
Grafted, Primed hNSCs Predominantly Differentiate Into Cells of Neuronal Lineage in the Injured Mouse Brain
To enhance the survivability of hNSCs through the grafting procedure, we injected cells rostral and medial to the injury epicenter. GFP-labeled hNSCs were accumulated mainly in the injury site (Fig. 7A). We then examined the differentiation phenotypes of the grafted hNSCs in mouse brains. Previously we found that FHL-primed hNSCs differentiated into neurons in intact or injured rat brains after transplantation under immunosuppression, overcoming the inhibitory environmental cues23,29. To determine whether these cells have the same potential in the injured mouse brain without immunosuppression, we stained sections with antibodies for neurons or neuronal precursors (DCX) and astrocytes (GFAP). Figure 7 showed that a majority of the grafted hNSCs stained positive for DCX (Fig. 7A and B) and only a few cells stained positive for GFAP (Fig. 7C and D). These results indicate that hNSCs primed with FHL (see Materials and Methods) predominantly differentiated into cells of a neuronal lineage in the injured mouse brain.

Transplanted hNSCs preferentially differentiated into neurons in injured mouse brains. Primed hNSCs were labeled with green fluorescent protein (GFP) 5 days prior to transplantation. Brain sections were collected 6 days postgrafting and double stained with antibodies against GFP and neuronal marker doublecortin (DCX) or astrocytic marker glial fibrilary acidic protein (GFAP). DAPI (4′,6-diamidino-2-phenylindole), nuclear counterstain. Confocal images show that the majority of GFP-labeled cells surrounding the injury site are double labled with DCX (A, B), whereas only a few colabeld with GFAP (C, D). Scale bars: 20 μm.
Both M1 and M2 Microglia/Macrophages Are Involved in Phagocytosis of Grafted, Primed hNSCs
Without immunosuppression, the hNSC xenograft most likely elicits an immune response and thus is rejected by the host33,34. To determine the fate of hNSCs grafted into the injured mouse brain without immunosuppression, we tracked the grafted hNSCs with GFP labeling with or without a GFP antibody and dual labeled them with various markers for microglia/macrophages at both 1 and 6 days posttransplantation. One day after grafting, some GFP-labeled hNSCs were surrounded by Iba1- or arginase 1-labeled microglia/macrophages, with few, if any, double labeled cells (Fig. 8A and B). Six days after grafting, the majority of GFP+ cells in the injury site were found to be costained with Iba1 (data not shown). Dual labeling detected GFP+ hNSCs colabeled with one of the four M1 or M2 markers (Fig. 8C–J). Many of these duallabeled cells showed morphologies of phagocytic gitter cells (arrows and insets in Fig. 8D, F, H, and J), indicating that both M1 and M2 microglia were involved in phagocytosis of grafted hNSCs.
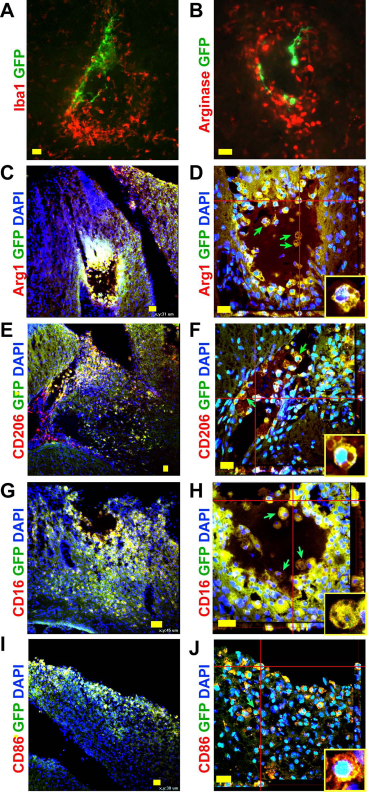
Phagocytosis of grafted hNSCs by microglia/macrophages in mouse brains. (A, B) Representative confocal images of grafted hNSCs (labeled with GFP in green) are surrounded by Iba1+ or arginase 1+ microglia/macrophages at 1 day posttransplantation. (C–J) Grafted cells surrounding the injury site are dual immunostained with GFP and M1 or M2 markers at 6 days postgrafting. M2 markers include arginase 1 (Arg1 in C, D) and CD206 (E, F); M1 markers are CD16/32 (G, H) and CD86 (I, J). (D, F, H, and J) Three-dimensional confocal images. Green arrows and insets, gitter cells. DAPI, nuclear counterstain. Scale bars: 20 μm (A, B, D–F, H, and J), 30 μm (C, I), and 45 μm (G).
Discussion
In this study, we investigated neuroprotective and immunomodulatory properties of transplanted hNSCs in mice with severe TBI. The main observations were that transplantation of primed hNSCs reduced axonal injury, accompanied by a diminished activation of brain microglia/macrophages and an altered ratio of M1 and M2 phenotypes. Meanwhile, we found that grafted hNSCs mainly differentiated into neurons, which were phagocytized by M1 or M2 microglia/macrophages 6 days posttransplantation.
Stem cell-mediated immunomodulation is an important mechanism underlying the neuroprotective effects of these cells. Two types of stem cells have been tested in animal models of neurotrauma: mesenchymal stem cells (MSCs) and NSCs. For brain injury, we and others have previously reported that intravenous administration of human or rodent bone marrow-derived MSCs (BM-MSCs) or multipotent progenitor cells suppressed microglial/macrophage activation and increased the M2/M1 ratio in injured rodent brains21,35,36, which was associated with long-term motor improvements after TBI 37 . The antineuroinflammatory effect of intravenously delivered stem/progenitor cells was suggested to be due to their ability to stimulate regulatory T cells in the spleen, which induces a systemic increase of circulating anti-inflammatory cytokines and an alteration of the locoregional milieu of the CNS. The altered intracerebral microenvironment then leads to modulation of the microglial/macrophage polarities in the brain. Besides this systemic immunomodulation, intracerebroventricularly administered MSCs also directly affect microglia/macrophages and lead them toward the M2 anti-inflammatory polarization 38 . Here we discovered that a different type of stem cell, hNSCs, when grafted intracerebrally, could also attenuate microglial/macrophage activation and favored a transition of these cells toward the M2 anti-inflammatory phenotype. The attractive feature of hNSCs, when compared to MSCs, is their unique nature of being committed to differentiation into neural lineage cells. Thus, hNSC transplantation may provide several layers of benefits as a potential therapeutic strategy for brain injury by modulating the microenvironment postinjury and replacing lost neural cells.
The immunomodulatory and associated neuroprotective effects of stem cells have also been reported in studies in which animals suffered spinal cord injury (SCI) and subsequently received intraspinal transplantation of NSCs or MSCs39,40. In the injured spinal cords, M1- and M2-associated genes were rapidly induced; M2 gene expression was transient and in most cases returned to preinjury levels by 48 h postinjury. In contrast, M1 gene expression was maintained for up to 1 month after injury. Collectively, these changes lead to an increase of the M1/M2 ratio by 7 days postinjury 16 . In our work, we chose the 7-day time point to assess the immune status of the injured mouse cortex with and without hNSC transplantation and confirmed a reversed M1/M2 ratio after cell grafting. Thus, we and others clearly demonstrate a transition of microglia/macrophages from the proinflammatory M1 to the anti-inflammatory M2 status and provide strong evidence for a common mechanism for stem cell-mediated neuroprotection independent of the types of stem cells, the route of administration, or the location of traumatic injury.
The neuroprotective effects of stem cell-mediated immunomodulation may include enhancing neuronal survival as well as reducing axonal injury. The latter has been extensively demonstrated in animal models of multiple sclerosis after grafting various types of stem cells41–43. TBI results in axonal injury, and both our previous and current studies show that hNSC transplantation dramatically reduces traumatic axonal injury. Although we have suggested previously that NSC-secreted glial cell line-derived neurotrophic factor (GDNF) may be responsible for axonal protection and regeneration 24 , it is possible that inhibiting microglia/macrophage activation and modifying their polarization may be another factor for NSC-mediated axonal preservation. In this regard, we note that neuroinflammation, particularly M1 proinflammatory activity, has recently been found to play a key role in axonal injury, whereas enhancing the M2 function promoted axonal regrowth after SCI16,44. It remains to be determined, however, to what extent GDNF or anti-inflammatory mechanisms are responsible for hNSC transplantation-mediated axonal protection and the relationship between NSC-secreted GDNF and NSC-mediated anti-inflammatory activity.
The stem cell-mediated M1-to-M2 transition is most likely due to promoting anti-inflammatory cytokines while reducing proinflammatory cytokines. We know that NSCs in the intact rodent brain have secretory protein profiles distinct from other brain cells, and these profiles have been suggested to be responsible for cytokine modulation 45 . In the present study, we found that hNSC grafts significantly reduced the level of TBI-induced proinflammatory cytokine receptor, IFN-γRβ, and increased the anti-inflammatory cytokine receptor, IL-4Rα. A lower density of microglia/macrophages with reduced levels of proinflammatory cytokines and increased anti-inflammatory cytokines has also been reported in animals intravenously administrated MSCs 36 . Several mechanisms underlying the MSC-mediated M1-to-M2 transition have been suggested, including modulating the expression of the neuroprotective CX3CR1 receptor on microglia through releasing CX3CL141,46, expressing an IL-1 receptor antagonist, reducing inflammatory nuclear factor kappa B (NF-κB) signaling, and secreting anti-inflammatory prostaglandin E2 and anti-reactive oxygen species (ROS) protein 47 . It remains to be determined whether NSCs grafted into a traumatic environment modulate microglia/macrophage-mediated inflammation through some or all of these same mechanisms.
It is possible that we may have underestimated the hNSC-mediated changes in M1 and M2 phenotypes, cytokine receptors, as well as APP and α-SMA. This is because Western blot analyses were performed on proteins collected from the entire injured hemisphere, whereas most M1 and M2 microglia accumulated only around the injury epicenter. Thus, it is possible to detect higher changes of those microglial markers and cytokine receptors in proteins from just the injured region. Furthermore, it will be interesting to determine how cytokines, cytokine receptors, and microglia in regions distal to the damage center respond to traumatic injury and cell grafting.
Microglia/macrophages serve critical roles in the maintenance of tissue integrity, development, and homeostasis. The process of phagocytosis is essential to M1- and M2-specific functions, and differentially polarized macrophages upregulate distinct machineries for the engulfment of specific phagocytic targets 48 . It has long been appreciated that M1 macrophages display an enhanced capacity for elimination of pathogens, whereas homeostatic functions such as phagocytizing apoptotic cells are mainly carried out by the M2 or M2-like macrophages48,49. However, it is unclear whether both microglial/macrophage phenotypes are involved in phagocytosis of grafted stem cells. In the present study, we transplanted hNSCs into injured mouse brains without administering immunosuppressants to observe the interaction between microglia/macrophages and xenogeneically grafted hNSCs. We found, for the first time, that the majority of grafted hNSCs, particularly those near the injury site, were engulfed by M1 or M2 microglia/macrophages. This indicates that both phenotypes are involved in phagocytosis on the sixth day posttransplantation. Interestingly, the majority of the phagocytic microglia/macrophages were double labeled with a neuronal marker, indicating that most of the grafted hNSCs differentiated into neurons before phagocytosis. Other groups, however, have previously reported that the microenvironment of the injured brain was unfavorable for neuronal differentiation of grafted hNSCs50,51. The discrepancy in neuronal differentiation is most likely attributable to the priming of hNSCs toward the neuronal lineage prior to transplantation in our protocol. Furthermore, because rejection of cellular implants usually happens within 2–4 weeks, additional temporal studies are needed to define the fate of grafted hNSCs and their interaction with two phenotypes of microglia/macrophages. It also remains to be determined whether immunosuppression, which is often used in NSC transplantation clinically, affects stem cell-mediated immunomodulation.
Conclusions
This study confirms the beneficial effect of hNSC transplantation for TBI and suggests that primed hNSCs have the ability to attenuate microglial/macrophage activation, leading them toward preferential differentiation into the neuroprotective M2 phenotype, which likely contributes to the neural repair effect of grafted hNSCs. The observed reduction of traumatic axonal injury in TBI mouse brains may be associated with an anti-inflammatory microglial polarization. Interestingly, an hNSC xenograft is short lived without immunosuppression, but they still attenuate inflammatory responses. As a therapeutic goal to amplify the neuroprotective effect of changes in microglia, further studies are needed to clarify how NSCs modulate this microglial/macrophage polarization.
Footnotes
Acknowledgments
The work was supported by grants from the TIRR Foundation Mission Connect to P.W., C.S.C., and R.J.G.; the Coalition for Brain Injury Research to P.W.; the Gilson Longenbaugh Foundation to P.W.; the Moody Project to P.W., D.S.D., and D.S.P.; and the J. S. Dunn Foundation to P.W. The authors thank Dr. Yongjia Yu and Dr. Richard Coggeshall for the critical reading of the manuscript. The authors declare no conflicts of interest.