
Abstract
Dental pulp stem cells (DPSCs) are reported as sources of mesenchymal stem cells (MSCs). MSCs are used as cell therapy options for various diseases. The present study examined the healing effects of DPSC injection on damaged bladder tissue in a chemically induced cystitis rat model. Cystitis was induced by hydrochloride injection into the bladder of female F344/NSlc rats. On the following day, DPSCs suspended in phosphate-buffered saline (PBS) were injected into the bladder and maintained for 1 h (DPSC injection group), while PBS alone was injected as the standard for comparison (PBS injection group). After 2 days following injection, considerable submucosal edema, vascular structure destruction, hemorrhage, and inflammatory cell invasion were observed both in the DPSC and PBS injection groups, with no difference in their degree of submucosal edema and hemorrhage. Six days after injection, vascular structure regeneration was observed in both groups; however, unlike the DPSC injection group, the PBS injection group showed traces of submucosal edema and hemorrhage. These results correlated with tissue concentrations of myeloperoxidase (MPO) and the inflammatory cytokines IL-1β, IL-6, and TNF-α. Furthermore, the intercontraction interval was prolonged, and the frequency of nociceptive behaviors was reduced in the DPSC injection group compared with the PBS injection group. DPSCs were found on the bladder epithelium until day 3 after injection. In the DPSC-conditioned media (CM), the trophic factors FGF-2, VEGF, and the C-C and C-X-C families of chemokines were detected. The results of DPSC injection into the cystitis rat model suggested that the injected cells promote the healing of the damaged bladder tissue by exerting various trophic effects while localizing on the bladder epithelium and that MSC injection is a potential novel therapy for interstitial cystitis/painful bladder syndrome.
Introduction
Symptoms of interstitial cystitis/painful bladder syndrome include pollakiuria and pain due to retention (7). The International Continence Society defined painful bladder syndrome as “the complaint of suprapubic pain related to bladder filling, accompanied by other symptoms such as increased daytime and nighttime frequency, in the absence of proven urinary infection or other obvious pathology” (1). An estimated 2.7% of US women aged 18 years or older (2) and 1.3% of US men aged 30–79 years (11) suffer from pain associated with interstitial cystitis/painful bladder syndrome. The underlying cause of interstitial cystitis/painful bladder syndrome remains unclear. However, the condition often accompanies damage to the bladder epithelium due to chronic inflammation (10), and the loss of bladder epithelial function leading to tissue destruction has also been reported (14). Although several clinically feasible therapies have been proposed, most of them provide relief for subjective symptoms through systemic drug administration (7). Because the direct administration of drugs into the bladder through the urinary tract is possible, serious or intractable cases can be locally treated. However, for some patients, such treatments have no effect.
Mesenchymal stem cell (MSC) transplantation has been reported to be an effective treatment for various diseases (17). Several MSC sources for cellular transplantation have been reported (3), one being dental pulp stem cells (DPSCs) (16). DPSCs have been shown to be a more efficient source of induced pluripotent stem cells (iPSCs) than skin cells, and these cells are now being banked in Japan (22). Dental pulp can be obtained from wisdom teeth that are not medically necessary; thus, there are few ethical concerns associated with their use. DPSCs are thus good candidates for cell therapy. In this study, we investigated the effects of DPSC injection on damaged bladder tissue.
Materials and Methods
This study was approved by the ethics committees and the animal care and use committees of the National Center for Geriatrics and Gerontology, Nagoya University and Aichi Gakuin University. All experiments were performed in accordance with the strict guidelines of the gene recombination experiment safety committee.
Cell Isolation and Culture Conditions
Normal human third molars were collected from adults (19–27 years, male, n = 6) at the Aichi Gakuin University Dental Hospital under the approved guidelines set by the School of Dentistry, Aichi Gakuin University. Pulp tissues were obtained from wisdom teeth that were not medically necessary. Research protocols that used DPSCs as materials were reviewed and approved by the ethical committee of the National Center for Geriatrics and Gerontology, Nagoya University and Aichi Gakuin University, where the study was performed according to the ethical codes of the Ministry of Health, Labour and Welfare of Japan. A written informed consent, approved by the above-mentioned ethical committees, was obtained from all enrolled subjects before participation in the study. The patients did not pay for their inclusion or treatment in this study. The dental pulp cells were enzymatically isolated from human dental pulp tissue using a previously described method (13). DPSCs cultured in Dulbecco's modified Eagle medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS; AusGenex, Loganholme, QLD, Australia) were detached at 60–70% confluence by incubation with 0.05% trypsin–EDTA (Life Technologies, Carlsbad, CA, USA) and subcultured at a 1:4 dilution under the same conditions. In this study, all cells were cultured in a humidified 37°C, 5% CO2 incubator.
Rat adipose stromal cells (ASCs) were prepared as a negative control for in situ hybridization. They were enzymatically isolated from control group rat subcutaneous abdominal fat and cultured by the same method as described for DPSCs.
Preparation of CM
The culture media were switched to DMEM without serum at 60–70% confluence for DPSCs at the fourth to fifth passage, and the CM were collected 24 h later and concentrated approximately 20-fold by an Amicon Ultra-15 centrifugal filter unit with an Ultracel-3 membrane (Millipore, Billerica, MA, USA). The concentrated CM were placed in a 96-well plate for analysis of cytokine and chemokine expression (Milliplex®MAP; human Cytokine/Chemokines Panel-Premixed 41 Plex; Millipore), in accordance with the manufacturer's guidelines. The secreted factors were quantified using a Bio-Plex 200 reader and Bio-Plex Pro wash stations (Bio-Rad, Hercules, CA, USA). Data of the secreted factors were analyzed with Bio-Plex Manager Version 3.0 software. All data were normalized to pg/ml/106. To evaluate the cytokines secreted by the DPSCs into the CM, comparisons were made with the CM of human adult dermal fibroblasts (NHDFs, Cat. No. CC-2511; Lonza, Basel, Switzerland) prepared in a similar manner.
Flow Cytometric Analysis
The DPSCs were characterized at the fourth to fifth passage. They were immunolabeled for 60 min at 4°C with a mouse IgG1 negative control (diluted 1:10) (FITC) (MOPC-21) (BioLegend, San Diego, CA, USA), a mouse IgG2 negative control (diluted 1:10) (FITC) (sc-2856) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), a rat IgG2b negative control (diluted 1:10) (PE-Cy7) (RTK4530) (BioLegend), a mouse IgG1 negative control (diluted 1:10) (PE) (MCA928PE) (AbD Serotec Ltd., Oxford, UK), and antibodies against CD105 (diluted 1:10) (FITC) (MEM-229) (Abcam, Cambridge, UK), CD29 (diluted 1:20) (FITC) (TS2/16) (eBioscience, San Diego, CA, USA), CD44 (diluted 1:20) (PE-Cy7) (IM7) (eBioscience), CD31 (diluted 1:10) (PE) (WM59) (BD Biosciences, San Jose, CA, USA), and CD45 (diluted 1:10) (APC) (HI30) (BioLegend). All antibodies were diluted in Hank's balanced salt solution (HBSS; Sigma-Aldrich) supplemented with 2% FBS (AusGenex) and HEPES (1:100) (Life Technologies). Flow cytometric analysis was performed using a FACSAria flow cytometer (BD Biosciences). The data were further analyzed, and graph overlays were created using FlowJo software (version 7.6.5; Tree Star Inc., Ashland, OR, USA).
Cystitis Model and Cell Injection
A rat model of chemically induced cystitis was developed using hydrochloride with a slight modification of our previous method (6). In brief, female F344/NSlc rats (n = 60; weight, 120–140 g) (Japan SLC, Hamamatsu, Japan) per time point were anesthetized with Escain (isoflurane; Mylan, Osaka, Japan). An indwelling catheter was inserted into the bladder of the rats through the urethral orifice. The lower abdomen was compressed to empty the bladder, into which 200 μl of 0.1 mol/L hydrochloride (Wako, Osaka, Japan) (n = 36) or 200 μl of saline for the controls (n = 12) was injected. The rats were then kept in the supine position for 1 min. The lower abdomen was again compressed to empty the bladder, which was then irrigated two times with 500 μl of saline. The catheter was removed and the rats recovered from the anesthesia. On the following day, all the rats were anesthetized, and an indwelling catheter was inserted into their bladders. The rats treated with 0.1 mol/L hydrochloride (Wako) were either injected with DPSCs (2.0 × 106 cells) suspended in 300 μl of PBS (Life Technologies) (DPSC injection group, n = 18) or 300 μl of PBS alone (PBS injection group, n = 18). The control rats were injected 300 μl of PBS alone in the bladder (n = 12). All rats were kept in the supine position for 1 h under anesthesia. The rats were divided into five groups: control (saline + PBS, n = 12), PBS day 2 (0.1 mol/L hydrochloride + PBS, n = 6), DPSCs day 2 (0.1 mol/L hydrochloride + DPSCs, n = 6), PBS day 6 (0.1 mol/L hydrochloride + PBS, n = 12), and DPSCs day 6 (0.1 mol/L hydrochloride + DPSCs, n = 12). After evaluation of these five groups, to investigate the length of time in which DPSCs are retained on the bladder urothelium, DPSCs days 1, 2, 3, and 4 group (0.1 mol/L hydrochloride + DPSCs, n = 3) were also added for in situ detection of DPSCs. All rats were immunosuppressed with a daily intraperitoneal infusion of 0.2 mg/kg/day tacrolimus (Prograf; Astellas Pharma, Tokyo, Japan), from 24 h before DPSC injection until the day of sacrifice. The time schedule for this experiment is shown in Figure 1. During the course of this experiment, catheter urine samples were collected from all the rat bladders for evaluation. Urine cultures were produced to exclude urinary tract infections with cysteine–lactose–electrolyte-deficient (CLED) agar (Nissui Pharmaceutical, Tokyo, Japan) in aerobic and anaerobic cultures at 37°C for 24 h. A 20-μl loop was used for the inoculation of urine samples. Samples were considered positive if they contained ≥105 CFU/ml. We confirmed that all samples were negative. The remainder of the urine samples was stored at −80°C until their use for cytokine quantification.
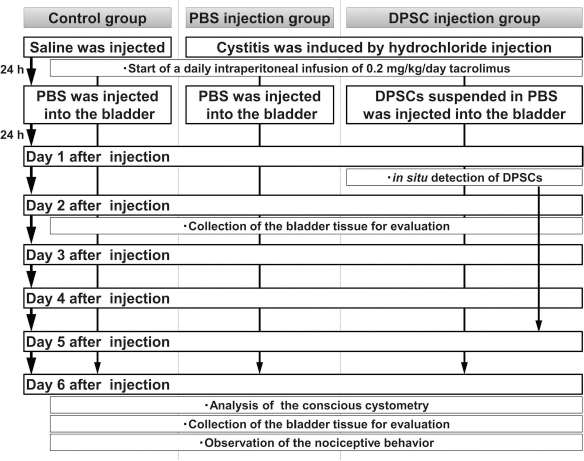
Time schedule for the animal experiment. Rats were divided into three groups: control, PBS injection, and DPSC injection. A rat model of chemically induced cystitis was developed using hydrochloride for both the PBS and DPSC injection groups, while saline was used for the control group. A daily intraperitoneal infusion of 0.2 mg/kg/day of tacrolimus started at the same time for all rats. After 24 h, either DPSCs or PBS was injected into the bladder. Then the time-based histological and physiological characteristics were analyzed to evaluate the effects of the DPSC injection.
Conscious Cystometry
Six rats per group were anesthetized with isoflurane. The bladder was exposed by a lower midline abdominal incision. PE-50 tubing (Clay Adams, Parsippany, NJ, USA) was inserted into the bladder through the dome. The tubing was pulled out through the abdominal incision, which was then tightly closed. After recovery from anesthesia, the rats were acclimatized for 2 h in a KN-326 Bollman-type restraining device (Natsume Seisakusho, Tokyo, Japan). Saline was infused at 0.04 ml per min for more than 1 h until the micturition interval stabilized. At least four reproducible micturition cycles were recorded. LabChart 7 software (AD Instruments, Oxford, UK) was used for data collection and analysis. The baseline pressure, voiding pressure threshold, peak voiding pressure, and intercontraction interval were evaluated.
Observation of Nociceptive Behavior
Six rats per group were placed in metabolic cages to acclimatize for at least 2 h, before being placed in a Ballman cage. PE-50 tubing (Clay Adams) was inserted into the bladder through the urethra, and residual urine was withdrawn. Resiniferatoxin (RTX; 300 μl, 3 mM) (LC Laboratories, Woburn, MA, USA) was infused into the bladder and maintained for 1 min to cause a sharp pain. The transurethral catheter was removed. Immediately after the rats were placed back in the metabolic cages (at about 10 to 15 s), a blinded observer recorded the incidence of licking and freezing for 15 min at 5-s intervals. Each incidence of licking or freezing during each 5-s interval was scored as one positive event (5).
Specimen Preparation and Protein Extraction
After cystometry, bladders were excised and divided in two parts (n = 6 in each group). One half of the bladder tissue was fixed in 4% paraformaldehyde (Nacalai Tesque, Kyoto, Japan) at 4°C overnight and embedded in paraffin wax (Sigma-Aldrich). Five-micrometer-thick paraffin sections were stained with hematoxylin–eosin (H&E) (Muto Pure Chemicals, Tokyo, Japan), Masson's trichrome (Muto Pure Chemicals) staining, Toluidine blue O (Amresco, Solon, OH, USA) staining, and immunofluorescence staining, and 10-μm-thick sections were used for in situ hybridization. The other half was homogenized in RIPA lysis buffer (Cell Signaling Technology, Beverly, MA, USA). The homogenate was centrifuged at 10,000 × g for 10 min, and the supernatants were stored at −80°C until use in the myeloperoxidase (MPO) assay and for cytokine quantification.
Immunofluorescence Staining
Immunofluorescence staining with cleaved caspase 3 was performed to detect apoptotic cells. Briefly, after deparaffinizing sections, the endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol for 30 min. To avoid nonspecific staining, the sections were pre-incubated in blocking solution (PBS containing 5% normal goat serum) for 30 min at room temperature, and primary antibody against cleaved caspase 3 (Asp175; Cell Signaling Technology) (1:300) was applied overnight at 4°C. After rinsing the sections, goat anti-rabbit IgG-Alexa Fluor 594 antibody (A-11012; Life Technologies) (1:200) was applied for 30 min at room temperature as secondary antibody. Finally, the sections were counterstained with 5 μg/ml Hoechst 33342 dye (Sigma-Aldrich).
MPO Assay
The MPO concentration was measured using an enzyme-linked immunosorbent assay kit (Hycult Biotechnology, Uden, Netherlands). The protein concentration was determined using a Pierce® BCA protein assay kit (Thermo Fisher Scientific, Pittsburgh, PA, USA). The MPO concentrations were standardized to tissue protein levels and were expressed as μg/mg.
Cytokine Quantification
Tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6 levels were determined using MAGPIX® with a MILLIPLEX®MAP Rat Cytokine/Chemokine Panel-Premixed 27 Plex (Millipore), in accordance with the manufacturer's guidelines. Quantification was performed on Bio-Plex 200 readers and Bio-Plex Pro wash stations (Bio-Rad) and was analyzed with Bio-Plex Manager Version 3.0 software. The cytokine concentrations in each tissue sample were standardized to tissue protein levels and expressed in pg/mg. The cytokine concentrations in each urine sample were standardized to urine creatinine levels and expressed in pg/mg. The creatinine levels in urine were measured using DetectX® urinary creatinine detection kits (Arbor Assays, Ann Arbor, MI, USA), following the manufacturer's guidelines. We confirmed whether quality control human cytokine (Millipore Cat. No. MXH6060) samples, which were positive controls for human cytokines/chemokines, worked in the rat panel. The cytokines/chemokines mentioned in this manuscript were not detected across animal species.
Human Y Chromosome-Specific Probe Preparation
Genomic DNA was extracted from human male DPSCs using a PureLink™ genomic DNA mini kit (Qiagen, Hilden, Germany); the human DYZ1-specific (Accession: KF941192) cDNA sequence was amplified by RT-PCR using primers (forward: 5′-CGAGCCCTTT CAATTTGAGTC-3′; reverse: 5′-GGAATGGAATGTC CTCTAATGG-3′) and AmpliTaq® Gold master mix (Life Technologies), and extracted with a QIAquick gel extraction kit (Qiagen) after electrophoresis. Purified cDNA was cloned in a pGEMT-easy vector system (Promega, Madison, WI, USA) and transformed into competent Escherichia coli cells (JM109; Promega). White and pale blue colonies were identified on X-Gal (final concentration: 50 mg/ml) (Nacalai Tesque) minimal agar plates and cultured in Luria–Bertani (LB) broth medium (Sigma-Aldrich) with ampicillin (final concentration: 100 μg/ml) (Sigma-Aldrich) for amplification. Plasmid DNA was purified from the E. coli cells using the Wizard® Plus Midipreps DNA purification system (Promega). After confirming the sequence of plasmid DNA, they were linearized with SpeI restriction endonuclease (Promega). Riboprobes were generated using the DIG RNA labeling mix (Roche Diagnostics, Pleasanton, CA, USA). Digoxigenin (DIG)-labeled riboprobes were purified as above and stored at −80°C until use.
In Situ Hybridization for Y Chromosome
Slides were deparaffinized in xylene (Kanto Chemical, Pulau Pinang, Malaysia) (two times) for 10 min. Endogenous peroxidase was inactivated by incubation with 0.3% hydrogen peroxide (Wako) in methanol (Kanto Chemical) for 30 min at room temperature. The tissue was digested with 20 μg/ml proteinase K solution (Life Technologies) in PBS for 15 min at room temperature. The slides were then denatured with 50% formamide (Kanto Chemical) in 4× sodium saline citrate (SSC; Takara Clontech, Kyoto, Japan) at 85°C for 10 min. The Y chromosome probe was denatured with 50% formamide (Kanto Chemical) in 4× SSC for 5 min at 75°C, added to the denatured tissue, covered with Parafilm® (Pechiney Plastic Packaging, Menasha, WI, USA), and incubated in a humid chamber overnight at 37°C. The Parafilm® were then removed in 4× SSC, and the slides were washed in 50% formamide (Kanto Chemical) in 2× SSC (Takara Clontech) for 1 h at 37°C and then in 0.1× SSC (Takara Clontech) for 2 h at 37°C. The slides were incubated with anti-digoxigenin-POD (Roche Diagnostics) and visualized using the TSA biotin system (PerkinElmer, Akron, OH, USA) and ImmPACT™ DAB peroxidase substrate (Vector Laboratories, Burlingame, CA, USA), following the manufacturer's guidelines. The sections were counterstained with hematoxylin (Muto Pure Chemicals).
Statistical Analyses
Data were expressed as means ± SD. The p values were calculated using Student's t-test and one-way analysis of variance (ANOVA) with Tukey's multiple comparison tests with SPSS 21.0 software (IBM, Armonk, NY, USA).
Results
Isolation and Evaluation of DPSCs
The morphology of the isolated DPSCs was stellate or spindle-shaped (Fig. 2A and B). The DPSCs exhibited proliferation potency (Fig. 2B). In the flow cytometric analysis, the DPSCs were positive for the expression of the stem cell markers CD105, CD29, and CD44 antigens and negative for expression of the endothelial cell marker CD31 antigen and the common lymphocyte marker CD45 antigen (Table 1, Fig. 2C-G).

Characteristics of dental pulp stem cells (DPSCs). (A) DPSCs at second passage on day 2 showed a stellate shape (arrow). (B) At the fifth passage, the self-replicating DPSCs showed spindle (triangle) and stellate (arrow) shapes. Flow cytometric analysis demonstrated that DPSCs were positive for (C) CD105, (D) CD29, and (E) CD44 antigens while negative for (F) CD31 and (G) CD45 antigens. Only representative examples are shown.
Flow Cytometric Analysis of Cell surface Markers on Dental Pulp Stem Cells (DPSCs)

Bladder Histological Findings Following Cell Injection
Considerable submucosal edema, vascular structure destruction, hemorrhage, and inflammatory cell invasion were observed in damaged bladder tissues compared with the controls when morphological evaluation was performed using H&E staining (Fig. 3). After 2 days following injection, there was no difference in the submucosal edema and hemorrhage between the DPSC injection group and the PBS injection group (Fig. 3B and C). After 6 days following injection, vascular structure regeneration was observed in all the groups; however, traces of submucosal edema and hemorrhage were found only in the PBS injection group (Fig. 3D and E). The PBS injection group showed a dense fibrosis that was located directly under the urothelium, while the image of the DPSC injection group is the same as that of the normal staining group (Fig. 4A). The infiltration of Toluidine blue-stained mast cells was only observed in the PBS injection group (Fig. 4B). The PBS injection group showed a higher expression of cleaved caspase 3 (apoptosis) than the DPSC injection group (Fig. 4C). The cleaved caspase 3 was mainly located over the bladder urothelium. The seriousness of the bladder inflammation correlated with the MPO concentrations (Fig. 5A), and the concentrations of the inflammatory cytokines IL-1β, IL-6, and TNF-α in tissues (Table 2) and urine (Table 3).

Inflammation following cell injection. Compared with the controls (A), considerable submucosal edema, vascular structure destruction, hemorrhage, and inflammatory cell invasion was observed at day 2 after injection in the PBS injection (B) and DPSC injection (C) groups, with no difference in recovery. Six days following injection, vascular structure regeneration was observed in the PBS injection (D) and DPSC injection (E) groups; however, submucosal edema was found only in the PBS injection group. Asterisks and arrows indicate submucosal edema and hemorrhage, respectively.

Treatment effects of DPSC injection. (A) Masson's trichrome staining. A dense fibrosis was observed directly below the urothelium in the PBS injection group, while an area of submucosal edema showed a low dense fibrosis (asterisks). The DPSC injection group appeared the same as the normal staining image. (B) Toluidine blue O staining. The infiltration of Toluidine blue-stained mast cells (arrows) was remarkably abrogated by DPSC injection. (C) Immunofluorescence staining for cleaved caspase 3 (red). There was a higher expression of cleaved caspase 3 (apoptosis) in the PBS injection group than in the DPSC injection group; this was mainly located over the bladder urothelium. The dotted line indicates the margin between the urothelium and stromal tissues.

Evaluation of the effects by dental pulp stem cell injection. (A) MPO assay revealed that MPO was higher in the PBS day 2, DPSC day 2, and PBS day 6 groups than in the DPSC day 6 group. (B) Cystometrograms showing representative intravesical pressure at day 6 after injection. Compared with controls (B-a), the intercontraction interval during the cystometrogram was significantly shorter in the PBS injection group (B-b). The DPSC injection group (B-c) had prolonged micturition intervals compared with the PBS injection group. (C) Nociceptive behavior was analyzed at day 6 after injection to evaluate licking (C-a) and freezing (C-b) incidence, which was more frequent in the PBS injection group than in the DPSC injection group. *p < 0.05 and **p < 0.01; #p < 0.05, ##p < 0.01 versus control.
Quantification of Proinflammatory Cytokines in the Bladder Tissue Using Rat Cytokine/Chemokine Panel

p < 0.05
p < 0.01 compared with the control;
p < 0.05 compared with the PBS day 6 group.
Quantification of Proinflammatory Cytokines in Urine Using the Rat Cytokine/Chemokine Panel
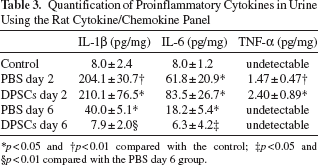
p < 0.05
p < 0.01 compared with the control
p < 0.05
p < 0.01 compared with the PBS day 6 group.
Conscious Cystometry
Excessive bladder activity (pollakiuria) was observed in the PBS injection group on day 6, as shown by the short bladder intercontraction intervals (Fig. 5B) (Table 4). In the DPSC injection group, the bladder intercontraction intervals on day 6 were shorter than normal values; however, they were significantly longer than those in the PBS injection group. Baseline and peak pressures were reduced to normal on day 6 in the DPSC injection group.
Cystometrogram Parameters

p < 0.05
p < 0.01 compared with the control
p < 0.05
p < 0.01 compared with the PBS day 6 group.
Analysis of Nociceptive Behavior
To evaluate responses to pain stimuli, the total instances of licking and freezing responses to chemical stimuli were counted. The frequency of licking (Fig. 5C-a) and freezing (Fig. 5C-b) were significantly lower for the DPSC injection group than for the PBS injection group at day 6.
Localization of DPSCs
To investigate the localization of the injected DPSCs within the bladder, human Y chromosome-specific probes were constructed, and in situ hybridization was performed. Human male DPSCs and ASCs were cultivated using the Chamber Slide™ system (Thermo Fisher Scientific), and in situ hybridization was performed on the slides on which the cells had adhered. The intranuclear labeling of human male DPSCs was confirmed (Fig. 6A). Rat ASCs were not labeled by the probes as a negative control (Fig. 6B). DPSCs in the bladder were identified on the bladder epithelium and were confirmed to be there until 3 days after injection (Fig. 6A-C).
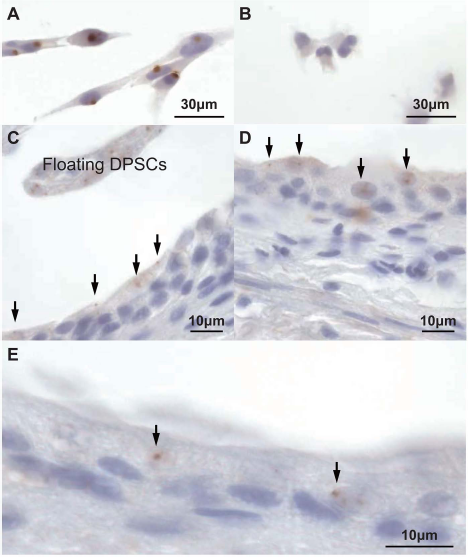
Detection of DPSCs using Y chromosome-specific probe. (A) DPSCs were labeled with a human Y chromosome-specific probe. (B) Rat-derived adipose stromal cells were not labeled. (C) The day after the injection, DPSCs were observed on the bladder epithelium and in the urinary bladder cavity as floating cells. On 2 (D) and 3 (E) days after injection, DPSCs were observed on the bladder epithelium. Arrows indicate DPSCs labeled by the human Y chromosome-specific probe.
DPSC-Secreted Cytokines
Compared with NHDFs, DPSCs released large amounts of more diverse cytokines (Table 5). Of note was the secretion of fibroblast growth factor 2 (FGF-2), vascular endothelial growth factor (VEGF), monocyte chemoattractant protein-1 (MCP-1) (CCL2), growth-related oncogene (GRO) (CXCL1, 2, 3), and fractalkine (CX3CL1).
Quantification of Cytokines Secreted by DPSCs and NHDFs Using the Human Cytokine/Chemokine Panel

CM, conditioned media; CCL, chemokine (C-C motif) ligand; CXCL, chemokine (C-X-C motif) ligand; CX3CL, chemokine (C-X3-C motif) ligand; MCP, monocyte chemoattractant protein; MDC, macrophage-derived chemokine; GRO, growth-related oncogene (GRO-α/CXCL1, GRO-β/CXCL2, GRO-γ/CXCL3); IP-10, interferon-inducible protein; PDGF, platelet-derived growth factor.
p < 0.05
p < 0.01 compared with NHDFs CM.
Discussion
The present study investigated the effects of DPSC injection on model rats with bladder tissue damage induced chemically via hydrochloric acid. Because hydrochloride causes mild homogeneous inflammation to the rat bladder, the hydrochloride-induced cystitis model (6) was used in this study.
Several clinical research groups have reported that inflammation, fibrosis, and mast cell infiltration are important in the pathophysiology of the interstitial cystitis/painful bladder syndrome (19,21). In the present work, in contrast to the PBS injection group, the DPSC injection group displayed histologically improved inflammation, with the damaged regions undergoing renewed healing on day 6 after injection. In the PBS injection group, a dense fibrosis was observed directly below the urothelium. The infiltration of mast cells was only present in the PBS injection group on day 6. The seriousness of the bladder inflammation correlated with the finding that the concentrations of MPO and proinflammatory cytokines (IL-1β, IL-6, and TNF-α) were reduced in samples extracted from bladder tissue in the DPSC injection group. Proinflammatory cytokine levels in urine samples were also reduced for this group. It is thought that the degree of healing of the damaged bladder tissue was reflected in these cytokine reductions. The proinflammatory cytokines IL-1β, IL-6, and TNF-α contribute to peripheral neuropathic pain and hyperalgesia (20). In the DPSC day 6 group, reduced tissue levels of these cytokines may have led to improved bladder compliance and a delayed micturition interval. However, the intercontraction interval time of the DPSC group at day 6 was significantly shorter than the control group in conscious cystometry. This result might reflect a declining compliance of the bladder, which was repaired by DPSC injection, compared with that of the control bladder. While hemorrhage and submucosal edema in the bladder epithelium were found in the tissues from the PBS day 6 group, the tissues from the DPSC day 6 group had recovered. Epithelial dysfunction of the bladder has been reported in interstitial cystitis (15); epithelial dysfunction leads to the migration of urinary solutes, particularly potassium, that depolarize nerves and muscles and can cause tissue injury (14). It has been reported by several clinical research groups that apoptosis is also important in the pathophysiology of interstitial cystitis/painful bladder syndrome (9,18,19). There was a higher expression of cleaved caspase 3 over the bladder urothelium in the PBS injection group than in the DPSC injection group. Compared with the PBS day 6 group, healing of the bladder epithelium was improved in the DPSC day 6 group, apparently leading to significant improvements in nociceptive behavior in response to chemical stimuli. It has been reported that immunosuppressive drugs such as cyclosporine and tacrolimus are effective to treat interstitial cystitis/painful bladder syndrome (8). Tacrolimus might soothe the pain of the inflammation in this study. However, because all rats were equally immunosuppressed with tacrolimus, tacrolimus probably did not interfere with the results of the different groups. Therefore, the difference of the nociceptive behavior frequency between the DPSC injection group and the control group may be attributable to the different response capability of repaired tissues and normal tissues to resiniferatoxin.
On investigating the localization of DPSCs in situ, the cells decreased in number over days, and they were localized on the bladder epithelium until day 3 after injection of the DPSCs. DPSCs were observed on the bladder epithelium and in the urinary bladder cavity as floating cells on the day after the injection of the DPSCs. At both 2 and 3 days after injection, DPSCs were observed on the bladder epithelium. The arrows in the figures indicate DPSCs that were labeled by a human Y chromosome-specific probe. The therapeutic effect of MSCs reportedly occurs through the secretion of trophic factors (3,4). Our previous research has demonstrated that CM isolated from DPSCs has a superior trophic effect and acts to promote migration and proliferation while suppressing apoptosis in bone marrow-derived stem cells (12). The comprehensive cytokine or chemokine quantitative analysis of CM isolated from DPSCs has not been conducted previously. Thus, to investigate the trophic factors released by DPSCs, the cytokines and chemokines in CM isolated from DPSCs were measured using a multiplex assay. It is thought that the trophic factors promoted the healing of damaged tissues because DPSCs secreted many kinds of cytokines or chemokines that could regulate immune reactions and angiogenesis. However, technical impediments in this work did not allow the simultaneous immunofluorescent staining for cytokines and DNA FISH for the Y chromosome. Therefore, one cannot assume that all the cytokines found in the supernatants of DPSC cultures are also present in the bladder tissue in vivo. The interaction analysis between each cytokine and in vivo molecules is a subject for future analysis. While the interstitial cystitis/painful bladder syndrome disease state has not been fully elucidated, the loss of bladder epithelium function and damage to bladder tissue has been reported (10,14). If the injection of MSCs such as DPSCs promotes tissue healing, then this may become a new therapeutic choice in the future.
Interstitial cystitis/painful bladder syndrome has been a model for the chronic disorder. However, the rat model of chemically induced cystitis utilized in this study indicated a state of acute inflammation and natural healing. Hence, the injection of DPSCs on the day after the induction of cystitis with hydrochloric acid may be too short to accurately model the interstitial cystitis/painful bladder syndrome. Therefore, investigations using a model that more accurately matches the interstitial cystitis/painful bladder syndrome are required. In addition, DPSCs were human derived, whereas the recipients were rats. Consequently, the limitations of the present study included the fact that it did not accurately reflect the chronic inflammation of the interstitial cystitis/painful bladder syndrome disease state and that the injected cells were heterologous. In clinical practice, autologous injection and comparison with other sources of MSCs will be necessary.
Footnotes
Acknowledgments
This work was supported by the Budget for promoting science and technology in Japan, which directly follows the policy of the Council for Science and Technology Policy (CSTP), chaired by the Prime Minister (M.N.), and the Research Grant for Longevity Sciences (23-10) from the Ministry of Health, Labour and Welfare (M.N.). Y.H. is a recipient of an Iwadare Scholarship from the Iwadare Educational Association for Dental Graduate Students. The authors would like to thank Enago and Dr. F. J. Alvarez for the English language review. The authors declare no conflicts of interest.