
Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of autoantibodies to components of the cell nucleus. These autoantibodies are predominantly produced with the help of follicular/helper T (Tfh) cells and form immune complexes that trigger widespread inflammatory damage, including nephritis. In recent studies, mesenchymal stem cells (MSCs) elicited diverse, even opposing, effects in experimental and clinical SLE. Here we investigated the effect of human bone marrow-derived MSCs (hBM-MSCs) in a murine model of SLE, the F1 hybrid between New Zealand Black and New Zealand White strains (NZB/W). We found that infusion of female NZB/W mice with hBM-MSCs attenuated glomerulonephritis; it also decreased levels of autoantibodies and the incidence of proteinuria and improved survival. These effects coincided with a decrease in Tfh cells and downstream components. Infiltration of long-lived plasma cells into the inflamed kidney was also reduced in the hBM-MSC-treated mice. Importantly, hBM-MSCs directly suppressed the in vitro differentiation of naive CD4+ T cells toward Tfh cells in a contact-dependent manner. These results suggest that MSCs attenuate lupus nephritis by suppressing the development of Tfh cells and the subsequent activation of humoral immune components. They thus reveal a novel mechanism by which MSCs regulate humoral autoimmune diseases such as SLE.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is a chronic, multigenic autoimmune disease characterized by a wide spectrum of clinical abnormalities, including skin rashes, arthritis, and nephropathy (25,27). Hallmark features of SLE include the production of autoantibodies (autoAbs) that recognize nuclear components, such as histone, DNA, and ribonucleoproteins. In particular, antibodies (Abs) against double-stranded DNA (dsDNA) are specific for SLE. AutoAbs to non-DNA antigens, including Sjögren syndrome A/Ro (SSA; a ribonuclear protein complex, also named Ro) and Sjögren syndrome B/La (SSB; an RNA-binding protein, also named La), were detected with lower specificity and prevalence than anti-dsDNA Abs (27). Immune complexes composed of these autoAbs are deposited in local tissues and trigger chronic inflammatory damage in multiorgan systems. In this way they act as key players in the development of symptoms of SLE.
The high-affinity pathogenic autoAbs originate mainly from self-reactive B cells as a result of the germinal center (GC) reaction in peripheral lymphoid tissues (17). Recognition of T-dependent self-antigens activates a fraction of naive CD4+ T cells to enter the developmental program of a helper T (Th) cell subset, named follicular helper T (Tfh) cells. These precursors of Tfh cells migrate into B-cell follicles via their CXCR5 activity in an early phase of the developmental program, and then complete the program leading to GC formation. The master regulator B-cell lymphoma (BCL)-6 is sufficient to instruct the precursors to commit to the Tfh cell fate, with expression of distinctive surface markers and signature cytokines, such as programmed cell death-1 (PD-1), inducible T-cell costimulator (ICOS), interleukin-21 (IL-21), and IL-4 (8,38). Tfh cells provide their cognate self-reactive B cells with the essential help needed to survive and to pass through tolerance checkpoints (4,41). In addition, the Tfh cells signal the B cells to undergo Ab class switching and affinity maturation, followed by differentiation into either memory B cells or plasma cells (PCs). Some of the PCs migrate into the bone marrow (BM) or inflamed tissues in search of survival niches. After arriving there, they undergo further maturation into long-lived PCs that are resistant to immunosuppressive treatments (26,33). Tfh cells are, therefore, crucial for the formation of affinity-matured, post-switched, long-lasting PCs.
Several lines of evidence point to the presence of dysregulated Tfh cells in both patients with SLE and models (9-11,18,20,21,31,39,45). In lupus-prone mice, an abundance of Tfh cells was shown to be closely linked to the excessive GC formation, high autoAb titers, and end-organ damage such as lupus nephritis (45). The breakdown of regulatory mechanisms that suppress the natural development of Tfh cells was shown to precipitate SLE-like symptoms (9,18,21). The finding that circulating precursors of Tfh cells were elevated in a subset of severe SLE patients led to these cells being proposed as disease markers (11,20,31,39). Taken together, these observations suggest that Tfh cells play an important role in the pathogenesis of SLE and that targeting them could be beneficial in opposing the initiation and/or progression of SLE.
Recent therapeutic approaches to SLE include infusion of mesenchymal stem cells (MSCs). These cells, derived mainly from BM, are multipotent with a capacity to differentiate into osteoblasts, chondrocytes, adipocytes, and myoblasts (32). They appear not to be effective as antigen-presenting cells (APCs), due to negligible levels of MHC molecules and costimulators on their surface. MSCs modulate the activities of diverse lymphoid-lineage cells, including T- and B-lymphocytes, natural killer cells, and dendritic cells, through contact-dependent engagement (e.g., via PD-1) and/or contact-independent mechanisms [e.g., via secreting indoleamine 2,3-dioxygenase (IDO), prostaglandin E2, IL-10, and transforming growth factor-β (TGF-β)] (2,23,28,30,42). The immunomodulatory potential of MSCs, along with their low immunogenicity, seems to offer a promising treatment for severe refractory autoimmune diseases. Indeed, allogeneic transplantation of MSCs derived from BM and umbilical cord blood conferred significant therapeutic effects on severe and refractory patients with SLE (36,43). The therapeutic benefit of MSC infusion was also observed in several experimental settings, although its efficacy and the source of the MSCs varied. In some studies, MSCs derived from human umbilical cord blood, adipose tissue, or BM were fully effective in preventing all SLE-associated abnormalities, such as autoAb production, proteinuria, and nephritis (5,6,12,47), while in another study, MSCs derived from mouse BM were effective in attenuating nephritis but not autoAb production and proteinuria (30). The suggested mechanisms underlying the efficacy of MSCs include suppression of B-cell activation and recovery of the balance between Th1 and Th2 cells or between Th17 cells and regulatory T cells (Tregs) (5,23,24,35,36). However, other studies have pointed to a detrimental effect of BM-derived MSCs, which enhanced B-cell activation and exacerbated symptoms of SLE (40,46). Moreover, despite the importance of Tfh cells in the pathogenesis of humoral autoimmune diseases, it is still not clear whether MSCs affect the activity of the pathogenic Tfh cells that arise in SLE.
Here we investigated the effects of human BM-derived MSCs (hBM-MSCs) in a murine model of SLE—the F1 hybrids between New Zealand Black (NZB) and New Zealand White (NZW) strains (referred to as NZB/W hereafter). These mice spontaneously produce antinuclear Abs at a level sufficient to trigger progressive immune complex-mediated glomerulonephritis and proteinuria (29). We found that infusion of female NZB/W mice with hBM-MSCs reduced SLE-associated abnormalities, and this was accompanied by a decrease in Tfh cells and downstream components of humoral immunity. The ability of MSCs to directly suppress the differentiation of naive CD4+ T cells toward Tfh cells was confirmed in vitro. Thus, our findings reveal a novel mechanism by which MSCs regulate the progression of humoral autoimmune disease.
Materials and Methods
Mice
Fifteen-week-old female NZB/W mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in a specific pathogen-free barrier facility at Hanyang University. Alternatively, 6-week-old NZB and NZW mice purchased from the Jackson Laboratory were crossed in our facility to obtain NZB/W mice. A total of 46 female NZB/W mice were used. Nine C57BL/6 and six OT II (ovalbumin peptide323-339-specific TCR transgenic) mice at 6 weeks of age were purchased from Orient Co. (Gyeonggi-do, Korea) and the Jackson Laboratory, respectively. The study was approved by the Institutional Animal Care and Use Committee.
Establishment of hBM-MSC Lines
MSCs derived from human BM were obtained from Corestem Inc. (Seoul, Korea). In brief, BM was aspirated from the posterior iliac crest of healthy donors, and mononuclear cells were collected by density gradient methods. The mononuclear cells were cultured in CSBM-A06 medium (Corestem Inc.) containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 2.5 mM L-glutamine and penicillin/streptomycin (WelGene, Gyeongsangbuk-do, Korea) in a 37°C 5% CO2 incubator for 3-5 passages. After washing out nonadherent cells, the adherent cells retained the canonical phenotype of MSCs (CD29+CD44+CD73+CD105+CD90+CD34-CD45-HLA-DR-) and were used in the experiments. The study was approved by the Institutional Review Board of Hanyang University Hospital.
Transplantation of hBM-MSCs to NZB/W Mice
Female NZB/W mice were infused with hBM-MSCs or vehicle (phosphate-buffered saline, PBS) at 1 × 106 cells/mouse/injection at 17, 19, and 21 weeks of age (n = 10-11/group) via retro-orbital injection of the venous sinus. In some experiments, 28-week-old female NZB/W mice were injected retro-orbitally with hBM-MSCs at 1 × 106 cells/mouse/injection five times at 1-week intervals (n = 8/group). All mice were sacrificed at 36 weeks of age for further analyses.
Enzyme-Linked Immunosorbent Assay (ELISA)
Sera were collected from NZB/W mice at 2- to 4-week intervals to measure the levels of Abs by ELISA, as described previously (16). In brief, to measure the levels of anti-dsDNA IgG, sera were diluted 1:5,000 in PBS (WelGene) and applied to immunosorbent plates (Nunc, Rochester, NY, USA) precoated with 5 μg/ml poly-L-lysine and 5 μg/ml thymic DNA (Sigma-Aldrich, St. Louis, MO, USA). A serum containing the highest titer of anti-dsDNA IgG was serially diluted and used as standard. The plates were incubated with anti-mouse IgG-biotin (Sigma-Aldrich) and streptavidin-HRP (BD Biosciences, San Jose, CA, USA). To measure the levels of anti-SSA/ Ro and anti-SSB/La Abs, sera were diluted 1:1,000 and assayed according to the manufacturer's instructions (Alpha Diagnostic, San Antonio, TX, USA).
To evaluate proteinuria, the concentration of albumin in urine was measured by quantitative ELISA using a mouse albumin ELISA kit (Bethyl, Montgomery, TX, USA), according to the manufacturer's instructions. Mice were scored as nephritic if the concentration of urinary albumin exceeded 100 mg/dl (5).
Histopathologic Examination of Lupus Nephritis
Kidneys and spleens were removed postmortem at 36 weeks of age, fixed in 4% paraformaldehyde (Sigma-Aldrich), embedded in paraffin, sectioned at 3 μm, and stained with periodic acid-Schiff (PAS) (Sigma-Aldrich) for kidneys, and hematoxylin and eosin (H&E) (Sigma-Aldrich) for spleens. The percentage of proliferating glomeruli and scores for lymphocytic infiltration were determined as described previously (30).
Fluorescence Microscopy
Mouse kidneys and spleens were removed postmortem at 36 weeks of age and embedded in OCT compound (Sakura Finetek, Torrance, CA, USA) and snap frozen in liquid nitrogen. Frozen sections were fixed in acetone (Sigma-Aldrich), blocked with 10% normal donkey serum (Sigma-Aldrich), and stained with 1:200 dilution of anti-IgG-biotin (Sigma-Aldrich) and 1:200 dilution of streptavidin-Cy3 (Invitrogen, Grand Island, NY, USA), 1:200 dilution of anti-C3-FITC (ICN Biomedicals, Irvine, CA, USA), 2 μg/ml anti-B220-allophycocyanin (eBioscience), 5 μg/ml anti-GL7-FITC (BD Biosciences), 1 μg/ml anti-CD4-Alexa Flour 647, and 1 μg/ml anti-B220-Alexa Flour 488 (Caltag Laboratories, Buckingham, MK, UK). Fluorescence images were acquired using a TCS SP5 confocal microscope (Leica, Ernst-Leitz-Strasse, Wetzlar, Germany). The area of GCs per field of 200x magnification was calculated using ImageJ software (NIH, Bethesda, MD, USA). GCs were classified as small (<7,000 μm2), medium (7,000-14,000 μm2), and large (>14,000 μm2).
Fluorescence-Activated Cell Sorting (FACS) Analysis
Single-cell suspensions of spleen and lymph node cells were prepared as previously described (16). To obtain mononuclear cells infiltrated in the kidneys, kidneys were removed from PBS-perfused mice, cut into pieces, digested with 500 μg/ml Liberase (Roche, Basel, Switzerland) at 37°C for 30 min, and strained through a 70-μm-pore cell strainer (SPL Life Sciences Co., Gyeonggi-Do, Korea). Cells were surface or intracellularly stained with an appropriate combination of monoclonal Abs (mAbs), as described previously (16). The mAbs and reagents used were anti-ICOS, anti-CXCR5, anti-CD4, anti-CD44, anti-PD-1, anti-B220, anti-CD138, anti-IL-4, anti-IL-17, anti-IFN-γ, anti-Foxp3, anti-CD25, anti-BCL-6, 7-aminoactinomycin D (7-AAD), and Annexin V [all from eBioscience (San Diego, CA, USA) or BD Biosciences]. They were used as FITC, PE, PerCP, allophycocyanin, or allophycocyanin-Cy7 conjugates.
Bromodeoxyuridine (BrdU) Incorporation Assays
Mice were fed drinking water containing 0.8 mg/ml BrdU (Sigma-Aldrich) for 14 days. Cells were extracted from spleens and kidneys, stained with anti-B220 and anti-CD138 mAbs, treated with 50 Kunitz units DNase (Sigma-Aldrich), and stained with anti-BrdU mAb (BD Biosciences). BrdU incorporation into B22010CD138+ cells was determined by FACS analysis.
In Vivo Tracking of MSCs
Superparamagnetic iron oxide (SPIO) nanoparticles coated with heparin (MW 12 kDa; Nanjing King-Friend Biochemical Pharmaceutical Co., Nanjing, China) were generated as described previously (referred to as SPIO-heparin) (15). hBM-MSCs were incubated with 25 μg/ml SPIO-heparin for 24 h at 37°C in a 5% CO2 incubator. The SPIO-heparin uptake efficiency of cells was assessed with Prussian blue (Sigma-Aldrich). Twenty-two-week-old NZB/W female mice were injected with 1.5 × 106 hBM-MSCs that had been labeled with SPIO-heparin and scanned by magnetic resonance imaging before and after infusion. Whole mice were scanned with a Philips (Amsterdam, Netherlands) 3.0T Ingenia with an 8-inch surface coil. Imaging sequences included T1-weighted spin echo and T2*-weighted gradient echo. The mice were killed on day 8 postinfusion, and organs were extracted. Genomic DNA was isolated using a DNeasy Blood and Tissue kit (Qiagen, Venlo, Netherlands) and assayed by quantitative PCR using primers for human β-actin (forward 5′-GAT CAT TGC TCC TCC TGA GC-3′ and reverse 5′-CAC CTT CAC CGT TCC AGT TT-3′) and mouse β2 microglobulin (forward 5′-TGA CCG GCC TGT ATG CTA TC-3′ and reverse 5′-TTT TGC GCT CAG GGA GTC TA-3′).
Lymphocyte Culture and Assays
Mouse splenic CD4+ T cells were sorted first by MACS (Miltenyi Biotec, Bergisch Gladbach, Germany) and then with a FACSARIAIII (BD Biosciences) to obtain naive CD4+ T cells (CD25-CD44+CD62L10) to >98% purity. CD4+ cells depleted of splenocytes by MACS were used as a source of APCs after 3,000 Rad γ-ray irradiation. hBM-MSCs were precultured in 24-well plates at 105 cells/ well overnight, and naive CD4+ T cells were added to the wells at 106 cells/well. To polarize the differentiation of the naive CD4+ T cells to Tfh cells, naive CD4+ T cells from C57BL/6 mice were stimulated with 1 μg/ml anti-CD3 mAb (eBioscience) together with an equal number of APCs in the absence (referred to as nonpolarizing or Th0 conditions) or presence of Tfh-polarizing conditions (10 μg/ml anti-IL-12 mAb, 10 μg/ml anti-IFN-γ mAb, 10 γg/ml anti-IL-4 mAb, 5 γg/ml anti-IL-2 mAb, 20 γg/ml anti-TGF-β mAb, 30 ng/ml recombinant IL-6, and 50 ng/ml recombinant IL-21; all from BD Biosciences; R&D Systems, Minneapolis, MN, USA; or eBioscience) (22). Transwells with 0.4-μm pores were used to physically separate the T cells from the hBM-MSCs, or alternatively, supernatants of hBM-MSCs cultures were added to the T-cell cultures, to 10% of the total culture volume. When naive CD4+ T cells sorted from OT II mice were used instead of those from C57BL/6 mice. 1 μg/ml anti-CD3 mAb was replaced by 1 μg/ml ovalbumin peptide323-339 (Peptron, Daejeon, Korea). The cells were cultured for 4 days and assayed by FACS to detect the markers of Tfh cells.
For proliferation assays, naive CD4+ T cells were labeled with 3 μM carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes, Grand Island, NY, USA) and stimulated with 1 μg/ml soluble anti-CD3 mAb and 1 μg/ml soluble anti-CD28 mAb (BD Biosciences) in the presence or absence of hBM-MSCs for 3 days, and the fluorescence intensity of CFSE was determined by FACS. To detect cytokine-producing Th cells, whole splenocytes were stimulated with 20 ng/ml PMA plus 1 μM ionomycin (Sigma-Aldrich) in the presence of Golgi-stop reagent (BD Biosciences) for 5 h, followed by intracellular FACS.
Naive B cells (B220+GL7-CD138-) from C57BL/6 mice were sorted by FACS to >98% purity, labeled with CFSE, and stimulated with 10 μg/ml lipopolysaccharide (LPS) (Sigma-Aldrich) plus 10 ng/ml recombinant murine IL-4 (BD Biosciences) in the presence or absence of hBM-MSCs in a ratio of 10:1. After 4 days of culture, the resultant cells were analyzed by FACS.
Statistical Analysis
Data are presented as mean ± SEM. Differences of Ab titer and histopathologic score between groups were evaluated by unpaired Student's t-tests. Differences of survival and incidence between groups were evaluated by Fisher's exact test. Values of p < 0.05 were considered statistically significant.
Results
Infusion of NZB/W Mice with hBM-MSCs Attenuates the Clinical and Histopathologic Manifestations of Lupus Nephritis
Previous studies of the effects of MSCs in NZB/W mice have given diverse or even contradictory results, depending on the experimental protocol and the source of MSCs (5,6,30,46). Therefore, we needed to test first whether the MSCs that we produced had a beneficial effect in NZB/W mice. We established lines of MSCs derived from healthy human BM (referred to as hBM-MSCs) and characterized the phenotypes by FACS analysis. The cells were scored as CD29+CD44+CD73+CD105+CD90+CD34-CD45-HLA-DR-, indicating that nearly all of them fit the criterion for canonical MSCs, as distinct from hematopoietic lineage cells (data not shown). Moreover, the cells could commit to adipogenic, osteogenic, and chondrogenic lineage fates under appropriate culture conditions (34,43), indicating that they retained the differentiation potential of MSCs (data not shown).
NZB/W mice were injected intravenously with hBM-MSCs or vehicle at 17, 19, and 21 weeks of age. Levels of anti-dsDNA Abs and urinary albumin rose progressively from 22 and 28 weeks of age, respectively, in the vehicle-treated mice. MSC infusion significantly increased these delay times to 30 to 34 weeks of age (Fig. 1A, B). Consistent with this, serum titers of anti-SSA/Ro and anti-SSB/La Abs were decreased (Fig. 1E, F). The cumulative incidence of proteinuria and survival were also significantly improved (Fig. 1C, D). Thus, hBM-MSCs administered during the preclinical phase suppress autoAb production and delay the onset and progression of lupus nephritis.

hBM-MSC infusion reduces autoantibody production and proteinuria in NZB/W mice. (A-F) Female NZB/W mice were injected intravenously with hBM-MSCs or vehicle at 17, 19, and 21 weeks of age (n = 10-11/group). (G and H) Twenty-eight-week-old female NZB/W mice were injected intravenously with hBM-MSCs 5 times at 1-week intervals (n = 8/group). (A and G) Serum levels of anti-dsDNA Abs determined by ELISA and displayed as mean ± SEM arbitrary units (AU)/ml. (B and H) Concentrations of urinary albumin determined by ELISA and displayed as mean ± SEM. (C and D) Cumulative incidence of proteinuria (>100 mg/dl) and survival rates. (E and F) Concentrations of anti-SSA/Ro and anti-SSB/La Abs in sera of 30-week-old mice determined by ELISA and displayed as mean ± SEM units (U)/ml. *p < 0.05, **p < 0.01, and ***p < 0.001 by Student's t-test (for A, B, E-H) and by Fisher's exact test (for C and D).
To determine whether the MSCs could inhibit the progression of already established lupus nephritis, we started to inject 28-week-old NZB/W mice that exhibited an intermediate severity of proteinuria and repeated it once a week for a total of 5 weeks. hBM-MSC infusion reduced the level of anti-dsDNA Abs, but did not alter the kinetics of urinary albumin accumulation (Fig. 1G, H). Therefore, this result demonstrated that hBM-MSC administration in the middle of the clinical phase could suppress auto Ab production, but was unable to reverse the progression of ongoing nephritis.
NZB/W mice that had been treated with hBM-MSCs or vehicle as in Figure 1A-F were examined histopathologically at 36 weeks of age. MSC infusion dramatically reduced splenomegaly and splenic hypercellularity (Fig. 2A). Deposition of immune complexes and complement factor C3 was prominent in the kidneys of vehicle-treated mice, but was not detected in those of the MSC-infused mice (Fig. 2B, C). In agreement with this, MSC infusion also reduced the percentage of proliferating glomeruli (Fig. 2D). Although the decrease in lymphocytic infiltration in PAS-stained tissues did not reach statistical significance, the fluorescence-labeled images clearly showed that infiltrates of CD4+ T cells and B220+ B cells were dramatically decreased in the kidneys of the MSC-infused mice (Fig. 2E). In addition to the kidneys, lymphoid aggregate foci composed mainly of CD4+ T cells and B220+ cells were reduced in the salivary glands of the MSC-infused mice, indicating that the MSCs were effective in suppressing sialadenitis (data not shown). We conclude that the ability of MSCs to prevent hyperactivation of immune responses in the spleen was associated with a reduction in immune complex-mediated glomerulonephritis and sialadenitis. We did not detect any adverse signs accompanying the hBM-MSC infusion, as judged by histopathologic examination of organs and by measurement of cellular and humoral immune responses (data not shown).

hBM-MSC infusion during the preclinical phase attenuates Histopathologic signs of lupus nephritis in NZB/W mice. Female NZB/W mice were injected intravenously with hBM-MSCs or vehicle at 17,19, and 21 weeks of age, and killed at 36 weeks of age. Spleens and kidneys were examined by histopathologic methods. (A) Photographs of spleens (left) and paraffin sections of spleens stained with H&E (right). (B, C, and E) Kidney cryosections stained with anti-IgG-biotin/streptavidin-Cy3, anti-C3-FITC, or anti-CD4-Alexa Fluor 647 plus anti-B220-Alexa Fluor 488, followed by immunofluorescence microscopy. (D) Kidney sections stained with PAS. Arrows indicate proliferating glomeruli. C57BL/6 (B6) mice were used as a negative control. Photographs are representative of four samples. Graphs show mean±SEM values of four samples. *p<0.05 by Student's t-test.
hBM-MSC Administration Reduces the Emergence of Immune Cells Mediating T-Dependent Ab Responses in NZB/W Mice
Tfh cells drive GC B cells to differentiate into PCs, and dysregulation of this process has been suggested to be responsible for lupus (9-11,18,20,21,31,39,45). Indeed, we found that 36-week-old female NZB/W mice displaying full-blown lupus nephritis contained more Tfh cells in their spleens, and circulating precursors of Tfh cells in their blood, than normal C57BL/6 mice (data not shown). The proportions of GC B cells and PCs, downstream targets of Tfh activity, also increased. Accordingly, GCs were numerous in the spleens of the NZB/W mice, but barely detected in the spleens of normal mice.
To examine which cellular components of humoral immunity were affected by hBM-MSC infusion, the cellular makeup of the spleens and blood of hBM-MSC- and vehicle-infused NZB/W mice was assayed by FACS. We found that hBM-MSC infusion decreased the proportion of Tfh cells (CD4+CXCR5+PD-1+) and their circulating precursors (CD4+CD44+CXCR5+PD-1+) (Fig. 3A, B). Reductions in the proportions of GC B cells (B220+GL7+) and PCs (B22010CD138+) were also evident (Fig. 3C, D). The total number of GCs in the spleens did not differ significantly between groups, but GCs of smaller size were more abundant in the hBM-MSC-infused mice, and those of larger size were correspondingly less abundant (Fig. 3E). These results, collectively, demonstrate that MSCs have the potential to suppress the emergence of Tfh cells and subsequent GC reactions in NZB/W mice.
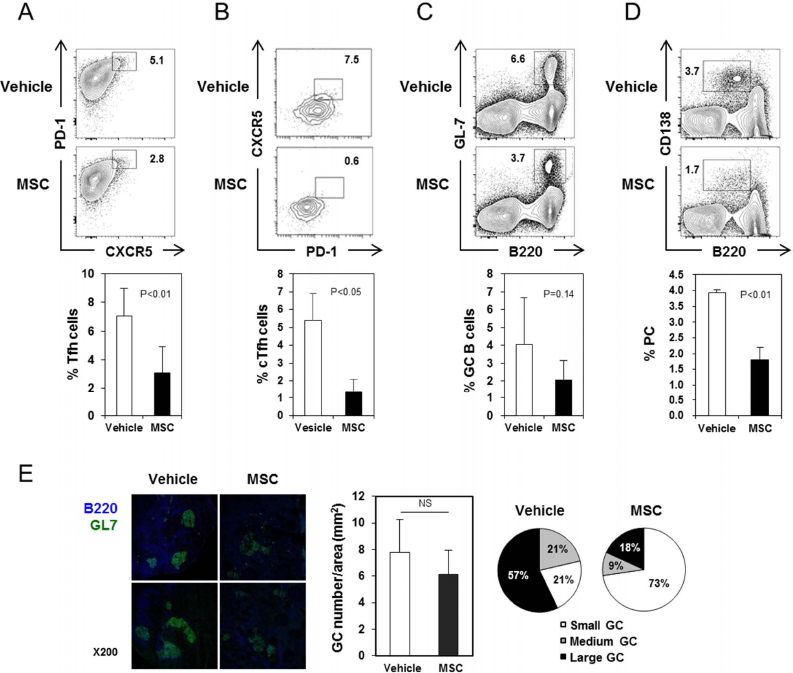
hBM-MSC administration reduces GC reactions in NZB/W mice. (A-D) Spleens and blood were removed from 36-week-old NZB/W mice that had been injected with hBM-MSCs or vehicle as in Figure 1A-F and assayed by FACS. (A) Representative FACS profiles of splenocytes (A, C, and D) and blood cells (B), gated on CD4+ cells (A), CD4+CD44+ cells (B), or whole live lymphocytes (C and D), with the percentages of cells within the indicated areas. The proportions of Tfh cells among CD4+ T cells, circulating Tfh (cTfh) cells among CD4+CD44+ T cells, GC B cells among whole live cells, and PCs among whole live cells are displayed as mean ± SEM (n=4-5/group). (E) Immunofluorescence staining of spleen cryosections for detecting GL7+ GC B cells (green) within B220+ B-cell follicles (blue). Representative images of four samples/group pooled from two mice/group are shown (left). GC numbers per mm2 are displayed as mean ± SEM (middle). GCs were classified into three categories depending on their sizes, and the percentages of GCs within each category are shown. NS, not significant.
MSCs have been shown to alter the balance between other Th subsets by suppressing the development of Th 1 and Th 17 cells and promoting the development of Tregs (5,12,24,35). In this regard, we examined the possibility that hBM-MSCs infused into NZB/W mice altered the frequency of Th subsets other than Tfh cells. We found that Th 1, Th2, and Th17 cells were quite rare in the NZB/W mice, and their proportions were not significantly altered by hBM-MSCs (Fig. 4). Moreover, hBM-MSCs did not increase the percentage of Foxp3+ Tregs. These results indicate that the disease-modulating effects of hBM-MSCs in NZB/W mice do not involve the activity of Th subsets other than Tfh cells.
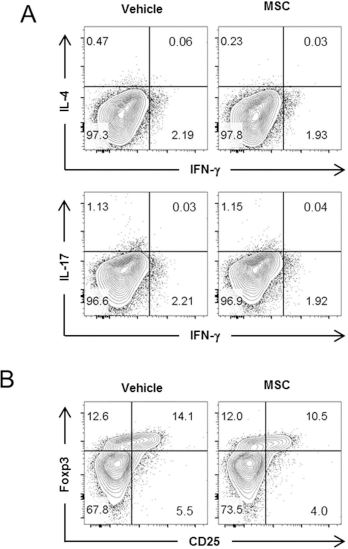
hBM-MSC infusion into NZB/W mice does not alter the proportions of Th subsets other than Tfh cells. Splenic CD4+ T cells isolated from NZB/W mice injected with hBM-MSCs or vehicle as in Figure 1A-F were assayed by intracellular FACS to detect cytokine-producing cells and Tregs. FACS profiles are representative of two independent experiments. (A) The percentages of IFN-γ-, IL-4-, or IL-17-producing cells among total CD4+ T cells. (B) FACS profiles with percentages of CD25+Foxp3+ Tregs among the whole CD4+ T cells.
hBM-MSC Infusion Reduces Infiltration of Long-Lived PCs Into the Kidneys of NZB/W Mice
PCs residing in local inflamed sites have been shown to survive for long periods until the inflammation is resolved (26). Moreover, long-lived PCs are less sus-ceptible to immunosuppressive treatments than conventional PCs (14), so that the emergence of these cells can render Ab-mediated diseases incurable. Accordingly, we found that NZB/W mice with severe lupus nephritis contained a substantial number of PCs (B22010CD138+) in their kidneys, whereas they were barely detected in normal C57BL/6 mice. Importantly, hBM-MSC infusion significantly reduced the numbers of PCs infiltrating the kidney (Fig. 5A). To test whether these renal PCs were indeed long-lived, NZB/W mice were fed BrdU for 14 days, and BrdU incorporation was assayed by FACS. BrdU-, nondividing, long-lived PCs comprised about 96% of renal PCs, but only 16% of splenic PCs (Fig. 5B). Given that the development of long-lived PCs requires the action of Tfh cells, it is understandable that the reduction in formation of Tfh cells brought about by MSCs should lead to presence of fewer long-lived PCs in inflamed tissues.
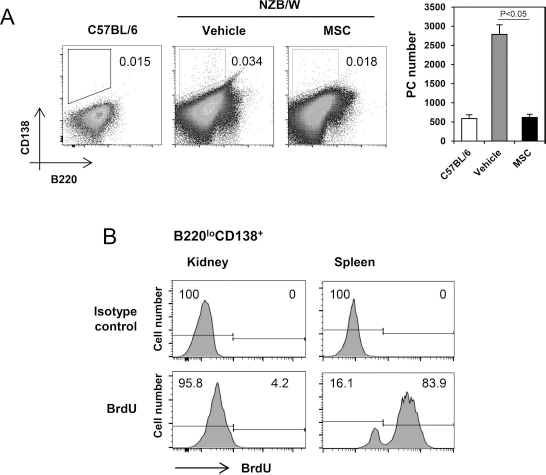
Transfer of hBM-MSCs to NZB/W mice reduces the number of long-lived PCs residing in the kidneys. Thirty-four-week-old NZB/W mice that had been injected with hBM-MSCs or their vehicle were fed with BrdU-containing water for 14 days. Single cell suspensions prepared from the kidneys and spleens were assayed by FACS. (A) FACS profiles gated on whole live cells with percentages of B22010CD138+ PCs are shown. The numbers of B22010CD138+ PCs a kidney are displayed as mean ± SEM. (B) FACS profiles gated on B22010CD138+ PCs with percentages of BrdU+ and BrdU- cells are shown. These figures are representative of three independent experiments.
Infused hBM-MSCs Home to Diverse Organs Including Lymphoid Organs
Our finding that MSCs reduce GC reactions in the spleen and lymph nodes suggested that these MSCs were trafficked to these organs. To track the infused cells, hBM-MSCs were incubated with SPIO-heparin, and iron staining showed that most of the cells were labeled with SPIO after 24-h incubation (Fig. 6A). The labeled MSCs were injected intravenously into NZB/W mice, which were examined by MR. The signal derived from the SPIO was most obvious in the spleen and liver 1 day postinjection and was sustained until day 4 postinjection (Fig. 6B).

Infused hBM-MSCs home to diverse organs. (A) hBM-MSCs were incubated with SPIO-heparin for 6 h or 24 h, and stained with Prussian blue. (B) Twenty-two-week-old NZB/W mice were injected intravenously with labeled hBM-MSCs and examined by MR. MR images obtained before (left) and 24 h (middle) or 96 h (right) after injection are shown. Arrowheads indicate the liver and spleen in the upper and lower panels, respectively. (C) On day 8 postinjection, organs were harvested postmortem, and real-time PCR of genomic DNA was conducted to quantify amounts of human β-actin DNA relative to mouse β2 microglobulin (mβ2M) DNA. Genomic DNA purified from untreated mice was used as a negative control.
On day 8 postinjection, genomic DNA was extracted from the organs of mice infused with MSCs, and their content of the human β-actin gene derived from hBM-MSCs was determined. The quantities of human β-actin gene relative to those of the mouse β-microglobulin gene were highly elevated in all the organs, other than lung and ovary, in the following order: liver, BM, spleen, kidney, lymph node, brain, and blood (Fig. 6C). These data indicate that MSCs homed systematically to diverse organs including the lymphoid organs and remained there for at least 8 days, a period presumably sufficient to modulate humoral immune responses.
hBM-MSCs Inhibit the In Vitro Differentiation of Tfh Cells
First, we tested whether hBM-MSCs could elicit immunosuppressive activity in mouse T cells in vitro. To this end, naive CD4+ T cells that had been purified from C57BL/6 mice and labeled with CFSE were cultured with anti-CD3 and anti-CD28 Abs in the presence or absence of hBM-MSCs. The addition of hBM-MSCs dramatically reduced the emergence of CFSEdim (i.e., dividing) CD4+ T cells, indicating that the hBM-MSCs behave as bona fide MSCs that can suppress the proliferation of mouse CD4+ T cells (Fig. 7A).

hBM-MSCs inhibit in vitro differentiation of Tfh cells. (A) Naive CD4+ T cells isolated from spleens of C57BL/6 mice were labeled with CFSE, and stimulated with anti-CD3 and anti-CD28 mAbs for 3 days in the presence or absence of hBM-MSCs. (B) Naive CD4+ T cells from C57BL/6 mice were labeled with CFSE and cultured for 4 days with anti-CD3 plus APCs in Th0 or Tfh conditions in the presence of vehicle control with hBM-MSCs or conditioned medium collected from MSC cultures (MSC-CM), or with hBM-MSCs in Transwells (TW). FACS profiles gated on CD4+CFSEdim cells with the percentage of BCL6+ICOS+Tfh cells are shown. (C) Naive CD4+ T cells from OT II mice were cultured with ovalbumin peptide323-339 plus APCs under Th0 or Tfh conditions with or without hBM-MSCs for 4 days. Histograms gated on CD4+ T cells with mean fluorescence intensities are shown. (D) Naive B cells (B220+GL7-CD138-) from C57BL/6 mice were labeled with CFSE and stimulated with LPS and IL-4 in the presence or absence of hBM-MSCs at a ratio of 10:1 cells. After 4 days of culture, cells were analyzed by FACS gated on B220+ or CD19+ B cells. Percentages of dividing CFSEdim cells among B220+ cells, of early (annexin V+ 7-AAD-) or late (annexin V+ 7-AAD+) apoptotic cells, and of CD138+ cells among dividing CFSEdim cells are shown. These figures are representative of more than three independent experiments.
Our in vivo data thus suggested that MSCs inhibit the differentiation of Tfh cells. To test this idea, naive CD4+ T cells purified from C57BL/6 mice were stimulated under either Th0- or Tfh-polarizing conditions in the presence and absence of hBM-MSCs, and the phenotypes of the resultant cells were analyzed by FACS. The Tfh-polarizing conditions were sufficient to instruct a fraction of naive CD4+ T cells to commit to the Tfh fate, as judged by the increased proportion of BCL6+ICOS+ cells. Addition of hBM-MSCs to the culture under the Tfh-polarizing conditions reduced the proportion of BCL6+ICOS+ cells among proliferating cells. This suppressive effect was not replicated by hBM-MSC-conditioned medium or when contact between MSCs and CD4+ T cells was prevented by Transwells (Fig. 7B). The hBM-MSC effect on the suppression of Tfh cell differentiation was independent of its suppression of T-cell proliferation because we analyzed only proliferating cells showing the CFSEdim phenotype. These results indicate that MSCs directly inhibit the differentiation of Tfh cells by contactdependent pathways.
To confirm the effect of MSCs on antigen-specific differentiation of Tfh cells, naive CD4+ T cells from OT II mice were stimulated with the cognate peptide and APCs under Th0 and Tfh conditions. hBM-MSC addition interfered with the induction of BCL6, ICOS, and PD-1 expression imposed by the Tfh conditions (Fig. 7C), demonstrating that MSCs inhibit the antigen-specific differentiation of Tfh cells.
Last, we tested whether hBM-MSCs directly affect the activation of B cells. Naive B cells undergo proliferation (CFSEdim), apoptosis (annexin V+), and differentiation to PCs (CD138+) in response to stimulation with LPS plus IL-4. Adding hBM-MSCs to such B-cell cultures did not significantly affect these three activities of B cells (Fig. 7D). Thus, the effect of MSCs on the suppression of autoAb production seems to depend on their action on CD4+ T cells rather than on B cells.
Discussion
SLE sometimes tends to be refractory to traditional treatments and to be life threatening, especially when major vital organs are invaded. In the present study, we demonstrated that lupus nephritis and sialadenitis that developed spontaneously in NZB/W mice were significantly attenuated by infusion of hBM-MSCs. This effect coincided with decreases in splenic Tfh cells, circulating Tfh precursors, GC B cells, and PCs, suggesting that the MSCs could suppress the in vivo development of pathogenic Tfh cells. Indeed, our in vitro experiments revealed that hBM-MSCs prevented naive CD4+ T cells not only from proliferating upon stimulation with anti-CD3 and anti-CD28 agonistic Abs but also from committing to the Tfh cell fate. The latter effect was not simply a consequence of the former, since it occurred in cells that had been proliferating in response to antigenic stimulation. The effect of MSCs on commitment was contact dependent and independent of soluble factors.
The identity of the surface molecule(s) that mediate the contact-dependent suppression of Tfh cell development by MSCs is not clear. We suspected the PD-1 ligand (PDL)/PD-1 system for a number of reasons. First, PDL was detected on the surface of MSCs and can signal T cells not to proliferate (2,30). Second, engagement of PD-1 on CD4+ T cells acts as a brake on Tfh cell differentiation (13). However we added anti-PDL1 neutralizing mAb to cocultures of CD4+ T cells with hBM-MSCs and found no effect (data not shown).
The existence of cross talk between MSCs and Tfh cells has been suggested previously. Xu et al. (44) found that the alleviation of Sjögren syndrome in NOD mice by allogeneic hBM-MSCs was accompanied by a decrease in the percentage of CXCR5+ cells in the CD4+ T-cell population and concluded that MSCs suppressed Tfh responses. However, there are at least two problems with this conclusion. First, although CXCR5 expression is a prerequisite for the development of Tfh cells, not all CXCR5-expressing CD4+ T cells appear to be Tfh cells because many T cells have been shown to express CXCR5 transiently upon activation (19). Moreover, the CD4+ T cells had been strongly stimulated in vitro to induce cytokine production, and that could have induced CXCR5 expression in a Tfh fate-independent manner. Second, the authors failed to demonstrate the decrease in PCs that should accompany a decrease in CXCR5+CD4+ T cells, if these cells are authentic Tfh cells. By contrast, we clearly demonstrated here that hBM-MSC-contacted CD4+ T cells were defective in the induction of a master regulator of Tfh cells, BCL6, as well as of Tfh-specific regulators such as ICOS and PD-1. Since circulating Tfh cells are a marker of active SLE, our finding that MSC infusion reduced the number of circulating Tfh cells further supports our conclusion that MSCs combat the emergence of Tfh cells in vivo.
One group has presented evidence that MSCs promote the survival of Tfh cells in follicular lymphomas and their conversion to follicular regulatory T (Tfr) cells (3), and they suggested that targeting MSCs could be a treatment for follicular lymphoma. Their finding was the opposite of that of the above-mentioned investigation (44) and also of ours, since we found that the MSCs we used supported neither the survival of any CD4+ T subsets nor the expansion of any Foxp3+ cell populations. This discrepancy is not surprising because MSCs are known to differ functionally depending on their microenvironment. For instance, MSCs are not constitutively immunosuppressive, but require a “licensing” step provided by proinflammatory molecules, such as interferon-γ (IFN-γ) and tumor necrosis factor-a (TNF-a) (28). We speculate that the lymphoma microenvironment, which contains elements different from those in autoimmune states, licenses MSCs to support the survival of Tfh cells.
The influence of MSCs on the physiology of B cells seems to be even more diverse than that on the physiology of T cells. Some studies have pointed to a potential for MSCs to suppress the survival and maturation of B cells in vivo and in vitro (1,7), while others showed, on the contrary, that MSCs actually augmented B-cell responses (37,40,46). For example, MSCs favored B-cell proliferation and/or differentiation into IgG-producing PCs in vitro (37,46). If that were also the case in vivo, MSC infusion might exacerbate humoral autoimmune disease. However, fortunately, our in vitro data indicate that the hBM-MSCs did not directly affect the activation of murine B cells, including their survival, proliferation, and differentiation into PCs. This implies that the effect of MSCs on the reduction of PCs observed by us was solely due to their impact on Tfh cells rather than on PC precursors. Therefore, our in vitro data regarding B cells further highlight the role of MSCs in the suppression of Tfh cells.
All the MSC effects on the suppression of GC responses imply that the MSCs infused intravenously were trafficked to lymphoid tissues. Indeed, we observed that hBM-MSCs were predominantly located in secondary lymphoid organs, such as spleen and lymph nodes. Besides lymphoid organs, we detected MSCs in most organs except ovary and lung on day 8 postinfusion. It is likely that the MSCs could not physically access the ovary, which is structured as an immune-privileged site. However, that is not the case for the lung, and we suspect that MSCs trafficked early to the lung might turnover rapidly and/or exit to other sites. It is interesting that MSCs were detected in the brain. This implies that the blood-brain barrier may be broken during the progression of SLE and suggests that MSCs might also be beneficial for treating SLE-associated neuropathy.
In summary, we have demonstrated here that infusion of hBM-MSCs slows the progression of lupus nephritis by suppressing the emergence of pathogenic Tfh cells. Our evidence for MSC targeting of Tfh cells includes the finding that hBM-MSCs suppress the in vitro differentiation of Tfh cells as well as in vivo autoAb production; furthermore, the MSCs have no effect on B cells and other Th subsets, and on the numbers of long-lived PCs in inflamed sites. Thus, our study has revealed a novel mechanism by which MSCs regulate humoral autoimmunity.
Footnotes
Acknowledgments
We thank Dr. H. S. Kim and I. Y. Chang for providing technical assistance and Dr. Julian Gross for editorial assistance. This work was supported by grants of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Korea (HI13C0016 and A120404). The authors declare no conflicts of interest.