
Abstract
Keratoconus is a corneal disorder characterized by a thinning of stromal tissue, and the affected patients have induced astigmatism and visual impairment. It is associated with a loss of corneal stromal keratocytes (CSKs). Hence, reconstructing stromal tissue with autologous CSK replacement can be a viable alternative to corneal transplantation, which is restricted by the global donor material shortage and graft rejection. Human CSKs are normally quiescent and express unique markers, like aldehyde dehydrogenases and keratocan. In serum culture, they proliferate, but lose their characteristic phenotype and become stromal fibroblasts. Here we report a novel culture cocktail to ex vivo propagate and maintain CSKs. Primary human CSKs were obtained from adult donors and cultured with soluble human amnion stromal extract (ASE), rho-associated coiled-coil-forming protein serine/threonine kinase inhibitor Y-27632, and insulin-like growth factor-1 (collectively named as ERI). Protein profiling using mass spectrometry followed by MetaCore” pathway analysis predicted that ASE proteins might participate in transforming growth factor-β (TGF-β) signaling and fibroblast development, cell adhesion, extracellular matrix remodeling, and immune response. In culture with 0.5% fetal bovine serum and ERI, the population of “activated keratocytes” was expanded. They had much lowered expression of both keratocyte and fibroblast markers, suppressed TGF-β-mediated Smad2/3 activation, and lacked fibroblast-mediated collagen contractibility. These “activated keratoctyes” could be propagated for six to eight passages ex vivo, and they regained CSK-specific dendritic morphology and gene marker expression, including aldehyde dehydrogenases, lumican, and keratocan biosynthesis, expression, and secretion when returned to serum-depleted ERI condition. This novel cocktail maintained human CSKs in both adherent and suspension cultures with proper keratocyte features and without the transformation to stromal fibroblasts. Thus, human CSKs can be ex vivo propagated as transient “activated keratocytes.” This could provide sufficient number of genuine CSKs for corneal tissue engineering.
Introduction
Keratoconus (KC, OMIM 14830) is a bilateral progressive corneal disease characterized by the stromal thinning, steepening, and collagen degradation (25). The corneal biomechanics are disrupted, and the cornea loses its shape and functions (transparency, refractivity, and strength), resulting in visual impairment. While KC is essentially a stromal disorder, there is evidence that the overlying corneal epithelium undergoes significant thinning (17). Besides the impact on vision and quality of life, KC has significant economic impact as it affects the young working age group. The disease onsets at puberty and progresses to middle age. In the US and many developed countries, it is one of the leading causes for corneal transplantation (including penetrating and lamellar keratoplasty); however, the worldwide donor shortage is a major issue restricting the success of these procedures (20,26). It is widely recognized that KC has a genetic basis with autosomal dominant inheritance, but is also associated with environmental cofactors (25). While the exact genetic basis is still undefined, the KC pathogenesis is postulated to involve significant keratocyte apoptosis triggered by inflammatory signaling, including interleukin-1-mediated Fas-ligand activation, and/or changes of tissue inhibitors of metalloproteinases (TIMPs)-1 and -3 (5,16,40). Reduced stromal keratocyte density and innervation are thus the major pathological features (33,46). In addition, altered stromal proteoglycan biogenesis due to keratocyte loss deregulates extracellular matrix (ECM) composition and disrupts cell adhesion, growth, migration, and apoptosis, thereby affecting the corneal integrity and transparency (28,57).
Cell transplantation to replace dead or diseased stromal cells by healthy corneal stromal keratocytes (CSKs) could be a viable alternative to conventional corneal transplantation (47). Human CSKs are quiescent in vivo and typically express stromal crystallins, including aldehyde dehydrogenases (ALDH, type 1A1 and 3A1), α-enolase, lactic dehydrogenase, and transketolase, contributing to corneal transparency (21). They also synthesize and deposit collagens and keratan sulfate proteoglycans (lumican, keratocan, and mimecan), as well as enzymes (such as collagenases) to degrade old matrix proteins for stromal matrix homeostasis. Human CSKs are expandable in serum-added culture; however, they exhibit a rapid loss of the aforementioned keratocyte phenotypes (7,13,49). The transient “activated keratocytes” differentiate into stromal fibroblasts and display a different gene expression profile (including fibronectin and metalloproteinases) (15,50). The synergistic action of serum and transforming growth factor-β (TGF-β) further transforms these cells to become myofibroblasts (19,23). Hence, the propagation of genuine human CSKs ex vivo has been problematic. While many studies have reported the proliferation of CSKs as stromal fibroblasts under serum culture, successful CSK cultivation under serum-free, supplement-defined, spheroid-generating conditions to maintain keratocyte properties has been documented with mouse (64), rabbit (3,14,18,22,24,30,34,35,39,61,62), and bovine cells (8,9,11,12,38,41–43,54). However, there is considerable variation in keratocyte properties between human and animal species (21), and these experimental results cannot be directly applicable for translational use in humans. The recent identification of adult human corneal stromal stem cells (hCSSCs) offers the opportunity for the development of functional keratocytes through population doublings (51). They produce stroma-like ECM components, but they are yet to be organized globally to produce functional stromal tissues (4,60). Lack of unique markers also makes the isolation of a homogenous and well-defined stem cell population difficult. In addition, human embryonic stem cell (hESC)-derived neural crest-like cells have also been induced to differentiate into keratocyte-like cells expressing keratocan and ALDH3A1 (1). However, the induction efficiency and cell purity are yet to be optimized. Therefore, expanding human CSKs ex vivo while maintaining their unique phenotypes is imperative and extremely desirable for their future application in cell transplantation and therapy.
Here we report a new culture method to propagate “activated keratocytes” from adult human corneal stromal tissue. These cells can regain appropriate keratocyte gene expression profile and functional keratocan biosynthesis. This novel protocol greatly enhances the efficiency of generating bona fide human CSKs for clinical and research use.
Materials and Methods
Human Corneal Stromal Tissues and Primary CSK Isolation and Culture
Research-grade cadaveric corneal tissues were purchased from Lions Eye Institute for Transplant and Research Inc. (Tampa, FL, USA). The age of the donor was 43.7 ± 17.6 years old, and the male-to-female ratio was 29:14. Corneoscleral specimens with endothelial cell density greater than 2,000 cells per mm2 were preserved in Optisol-GS (Bausch & Lomb Surgical, Irvine, CA, USA) and transported at 4°C to the laboratory. After washes with sterile PBS (Invitrogen, Carlsbad, CA, USA), the central button (8 mm in diameter) was trephined and treated with dispase II (20 mg/ml; Roche, Basal, Switzerland) followed by gentle scrapping to completely remove the corneal epithelium and endothelium. The stromal tissue fragments were digested with collagenase I (1 mg/ml; Worthington, Lakewood, NJ, USA) in keratocyte basal medium (KBM) for 12 h at 37°C. Single cells were suspended in KBM, which was DMEM/F12 medium (Invitrogen) supplemented with 2 mM l-glutamate, 20 mM HEPES, 1% insulin–transferrin–selenate (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 1% MEM Eagle's vitamin mix (Lonza, Basel, Switzerland), 1% MEM nonessential amino acids (Invitrogen), 1 mM l-ascorbate 2-phosphate (Sigma-Aldrich, St Louis, MO, USA), and antibiotics (penicillin S, streptomycin sulfate, and amphotericin B; Invitrogen). Cells were seeded at 104 cells per cm2 on collagen I-coated culture wells (BD Biosciences, Franklin Lakes, NJ, USA). To propagate genuine CSKs, our results must meet three basic requirements: (1) maintenance of cells with dendritic morphology, (2) lack of fibroblast features (negative expression of αSMA and F-actin stress fiber pattern), and (3) expression of CSK markers (ALDH1A1, ALDH3A1, lumican, keratocan, and COL8A2). We cultured the cells with various chemicals and growth factors, including soluble amnion stromal extract (ASE; preparation details as shown below), ROCK inhibitor Y27632 (10 mM; Millipore, Billerica, MA, USA), insulin-like growth factor 1 (IGF1; 10 ng/ml; Invitrogen), and/or fetal bovine serum (FBS; Invitrogen). Medium change was performed every 3 days. When cells reached about 60% confluence, they were subpassaged with TryPLE Express (Invitrogen) and plated to new collagen I-coated culture surface under the same seeding density. Cells not exceeding passage 6 were used in the experiments.
Preparation of Soluble Amnion Stromal Extract (ASE)
Fresh human fetal amnion (n = 3, fetuses of either gender) was isolated from placenta (mothers were younger than 40 years old) after cesarean section, with written consent and institutional review board-approved protocol. After rinses with sterile saline to remove blood traces, amniotic membrane (AM) was manually peeled from the chorion. The proximal AM close to the placenta was taken for ASE preparation. The tissue was cut into pieces (4 × 4 cm2 in size) and devitalized in DMEM (Invitrogen) containing 50% glycerol (Sigma-Aldrich) at −80°C. After PBS rinses, AM pieces were drip-dried, weighed, and ground under the air phase of liquid nitrogen. The homogenate was suspended in ice-cold sterile PBS (5 ml per gram tissue). All following steps for protein extraction were performed at 4°C. The suspension was rotated at 300 rpm (Rotator RS-24; Boeco, Hamburg, Germany) for 48 h and centrifuged at 25,000 × g for 20 min to remove insoluble debris. The supernatant was further spun with Ultra-centrifugal Filter (UltraCel-30k and/or −3k, Amicon™; Millipore) at 4,000 × g for 30 min. Various ASE fractions (refer to the Results section) were collected and stored at −80°C. Total protein concentration was determined by Protein DC assay (Bio-Rad, Hercules, CA, USA). TIMP1 was quantified by human TIMP1 enzyme-linked immunosorbant assay (ELISA) (Invitrogen). The protein sample was denatured in buffer containing 50 mM Tris-HCl (Bio-Rad), 2% SDS (Bio-Rad), 1% β-mecaptoethanol (Bio-Rad), 5% glycerol, and bromophenol blue (Sigma-Aldrich) and resolved using gradient SDS-PAGE (4–20%; Bio-Rad). Protein bands in gel were revealed by Coomassie blue (Sigma-Aldrich).
TGF-β-Induced Smad2/3 Expression
To assess whether ASE fractions affected TGF-β-induced changes of cell phenotype and Smad2/3 localization, human recombinant TGF-β1 (10 ng/ml; Invitrogen) was added to human CSKs in KBM with or without ASE fractions for 3 h and 3 days, respectively. The cells were fixed for immunofluorescence assay of Smad2/3 and F-actin expression (protocol details as shown below). Under fluorescence microscopy, minimums of 10 fields at 20× (objective) were obtained, and the percentage of cells displaying stress F-actin fiber pattern and percentage of cells with nuclear Smad2/3 expression were quantified, respectively. From triplicate experiments, the results were expressed as median and interquartile range (IQR), and statistical significance was determined using Kruskal–Wallis and Dunn–Bonferroni post hoc test for type I error p adjustment.
Immunofluorescence
Cells on collagen I-coated coverslips were fixed with neutral-buffered 2% paraformaldehyde (Sigma-Aldrich), quenched with ice-cold 50 mM ammonium chloride (Sigma-Aldrich) and permeabilized with 0.15% saponin (Sigma-Aldrich). After blocking with 1% bovine serum albumin (BSA; Sigma-Aldrich) and 2% normal goat serum (Invitrogen), they were incubated with primary antibodies (Table 1). The secondary antibodies were conjugated with either AlexaFluor 488 or Red-X (Jackson InnumoResearch Laboratories, West Grove, PA, USA). After washes, sections were mounted in Fluoroshield with 4′,6-diamidino-2-phenylindole (DAPI; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and viewed under fluorescence microscopy (Carl Zeiss, Oberkochen, Germany) or confocal laser-scanning microscopy (LSM 510 Meta; Carl Zeiss).
Primary Antibodies
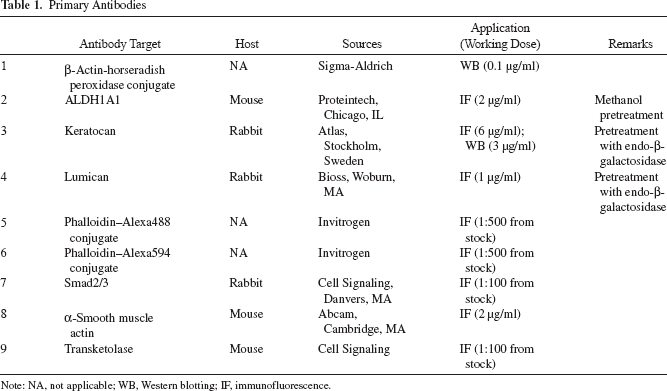
Note: NA, not applicable; WB, Western blotting; IF, immunofluorescence.
One-Dimensional Nanoscale Liquid Chromatography Coupled to Tandem Mass Spectrometry (Nano LC-MS/ MS) and Pathway Analysis
ASE samples (100 μg) were denatured with Tris-HCl (0.1 M), SDS (2%), and Tris-(2-carboxyethyl) phosphine (TCEP; 33 mM; Sigma-Aldrich) at 60°C for 1 h (59), followed by washing with urea (1 M.; Sigma-Aldrich), and blocking with iodoacetamide (Sigma-Aldrich) in urea in a centrifugal filter unit. The sample was collected by spinning at 14,000 × g for 10 min, washed with urea, and equilibrated with ammonium bicarbonate (50 mM; Sigma-Aldrich) before trypsin digestion. After washes, the digested sample was eluted, and trypsin was quenched by formic acid (10%; Sigma-Aldrich). It was then desalted with Silica C-18 ultra-microspin column (Nest Group, Southborough, MA, USA) and analyzed by one-dimensional nano LC-MS/MS using Dionex UltiMate 3000 (Thermo Fisher Scientific, Waltham, MA, USA) coupled with AB Sciex Triple TOF 5600. The operation details and proteomics data processing are described in Supplementary Methods (https://www.seri.com.sg/wp-content/uploads/2015/07/Supplementary-Materials_Dr-Gary-Yam.pdf). Pathway analysis was done by MetaCore™ software (GeneGO, San Diego, CA, USA). “Pathway Maps” were generated with p < 10−4 representing the pathways significantly enriched with the identified proteins.
Hanging Drop Culture
We employed a standard protocol of hanging drop method (10) with modifications to assess the cell growth in a three-dimensional ex vivo environment. Human CSKs (5,000 cells) at passage 5 were suspended in 100 μl medium, and 10-ml drops were applied to the inner lid side of a culture plate (60 mm diameter; Corning, Shanghai, China), and the plate reservoir was maintained with PBS to ensure humidity. The lid was placed back to the culture plate, and the setup was left at 37°C for 96 h. Under phase microscopy, the formation of cell sheet or aggregate was recorded. The amount of flattened cells or aggregates was quantified, and the median percentages were compared with the significance calculated by Kruskal–Wallis test and adjusted for type I error p value using Dunn-Bonferroni post hoc test.
Collagen Gel Contraction Assay
Human CSKs were suspended in bovine type I collagen solution (2.5 mg/ml, PureCol; Advanced BioMatrix, Poway, CA, USA) at a density of 105 cells/ml, and 0.5 ml mixture was added to each well of 24-well plate (Corning) coated with 1% BSA (Sigma-Aldrich). Collagen gel was formed at 37°C followed by being overlaid with medium. After 24 h, the gel was released from the culture well with a sterile needle to initiate contraction. At 48 h, the collagen gel was pictured, and gel size was measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/index.html). The percentage of gel size to the size of culture well was calculated and compared among treatments. The experiment was done in quadruplicate, and the percentages of gel contraction were represented as median and IQR. Intergroup significance was calculated by Mann–Whitney U-test. Multiple comparison was performed with the Kruskal– Wallis test and Dunn–Bonferroni correction.
Culture to Revert CSKs From “Activated Keratocytes”
Human CSKs were cultured in KBM added with 5 μg/ ml ASE, 10 mM Y27632, 10 ng/ml IGF1, and 0.5% FBS (termed as 0.5%SERI) for 3 or 7 days, followed by replenishing with medium of the same formulation except FBS (serum-free ERI) for another 3 or 7 days, respectively (schematic diagram shown in Fig. 1). Cells cultured uninterruptedly in ERI, 0.5%SERI, or KBM added with FBS served as controls. Fresh medium was replenished every 3 or 4 days. At days 6 or 14, cells and conditioned media were collected for immunofluorescence, RNA, and Western blot analyses.

A schematic diagram showing the media switch protocol. Human corneal stromal keratocytes (CSKs) at passage 5 were cultivated in 0.5%SERI for 3 or 7 days before the medium was switched to ERI without serum for another 3 or 7 days. Controls were cells cultured in ERI or 0.5%SERI for the whole length of 6 or 14 days. ^Regular medium change.
Quantitative PCR Analysis
Cells were collected in TRIzol (Invitrogen) and extracted with chloroform-isoamyl alcohol (Sigma-Aldrich) followed by RNeasy kit (Qiagen, Valencia, CA, USA) and on-column RNase-free DNase kit (Qiagen) according to the manufacturer's protocol. Reverse transcription of total RNA (1 μg) was performed with Superscript III RT-PCR kit (Invitrogen) using random hexanucleotide primer (10 ng/ml, Invitrogen) (Supplementary Table 1; https://www.seri.com.sg/wp-content/uploads/2015/07/Supplementary-Materials_Dr-Gary-Yam.pdf). Gene expression was assayed by quantitative realtime PCR (qPCR) using Sybr Green Supermix (Bio-Rad) in GFX96 Real-time System (Bio-Rad). Experiments were run in quadruplicate. Relative gene expression of each sample (ΔCT) was normalized by the mean CT value to house keeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (CTGAPDH) and expressed as median and IQR. Fold changes to human stromal tissue or untreated cells were calculated. Significance was determined by Kruskal-Wallis test with Dunn-Bonferroni correction.
Keratocan Expression
Conditioned medium was spun to remove cell debris. Cells were suspended at 105 cells/ml in PBS added with 0.5% Triton X-100 (Sigma-Aldrich) on ice for 20 min. After spinning at 25,000 × g for 15 min at 4°C, Triton X-100 insoluble fraction was collected and was further extracted in buffer containing 4 M guanidine-HCl (Sigma-Aldrich), 10 mM sodium acetate (Sigma-Aldrich), 10 mM disodium EDTA (Sigma-Aldrich), 5 mM aminobenzamidine (Sigma-Aldrich), and 0.1 M ∊-amino-n-caproic acid (Sigma-Aldrich), pH 7.2. Both samples were concentrated through an Amicon™ Ultra Centrifugal Filter (3k cutoff; Millipore) at 14,000 × g for 20 min at 4°C. Proteins were recovered in 0.1 M Tris acetate buffer (pH 6.0; Sigma-Aldrich) with 6 M urea, and the protein concentration was quantified at OD280. Protein aliquots (100 μg) were biotinylated using EZ-linked Sulfo-NHS-Biotinylation kit (Thermo Fisher Scientific, Waltham, MA, USA) under manufacturer's instruction. Briefly, 11 mM Sulfo-NHS-biotin in PBS was added in a 20-fold molar excess to sample proteins and incubated for 1 h at room temperature with rotation. The mixture was then allowed to absorb in a prewashed Zeba desalt spin column (Thermo Fisher Scientific) for 10 min and then centrifuged. The flow-through with biotinylated proteins was divided into two fractions. One was treated with 0.1 U/ml endo-β-galactosidase (Sigma-Aldrich) in phosphate buffer (pH 5.8) for 1 h at 37°C, and the untreated half was left on ice. Both fractions were immunoprecipitated with Protein A-conjugated magnetic beads (Millipore) prebound with polyclonal antibody against keratocan (Sigma-Aldrich) (Table 1). After magnetic separation and washes in PBS, the beads were denatured in 50 mM Tris-HCl (pH 6.8) added with 1% SDS and 0.25 M β-mercaptoethanol at 95°C. The protein sample was then resolved using gradient SDS-PAGE (4-20%) and Western blotted with a streptavidin-horseradish peroxidase-conjugated antibody (Thermo Fisher Scientific). Signal was detected with enhanced chemiluminescence (Clarity™; Bio-Rad). Band intensity was analyzed by Quantity One Imaging software (Bio-Rad) and the intracellular expression of keratocan was normalized with that of β-actin for comparison.
Results
The effect of soluble ASE on suppressing the fibroblast transition from human CSKs during ex vivo culture was first assessed. When resolved by 4-20% denatured gradient gel electrophoresis, Coomassie blue staining revealed that ASE contained about 50 major protein bands with molecular sizes ranging from 5 to 300 kDa. Using differential centrifugal filtration, we fractionated ASE according to the molecular size ranges. Fraction 1 (F1) was the original extract, F2 contained proteins ≥3 kDa, F3 with proteins ≥30 kDa, F4 with proteins at 3-30 kDa, F5 with proteins ≤3 kDa, and F6 had proteins ≤30 kDa. To examine which ASE fraction was effective in suppressing CSK transition to fibroblasts, we studied the TGF-β-mediated nuclear localization of Smad2/3. As shown in Figure 2, human CSKs at passage 5 plated on collagen I-coated culture surface (104 cells/cm2) were responsive differently to the addition of recombinant human TGF-β1 (10 ng/ml). Nuclear Smad2/3 was detected in 12.5% (8.9-14.6%) (median; IQR) cells without TGF-β1 but increased to 45.8% (39.1-51%) after 3 days of TGF-β challenge (20%; 18.3-26.7% when incubated for 3 h). When compared to the TGF-β1-treated group, incubation with whole ASE or various ASE fractions significantly suppressed nuclear Smad2/3 localization (p < 0.05; Kruskal-Wallis test with Dunn-Bonferroni correction), except for F5 (≤3 kDa). While most ASE fractions maintained a lower median percentage range (4-11%), F5 culture resulted in 29.4% (24.6-34.1%) and 31.6% (27.7-34.2%) cells having nuclear Smad2/3 at 3 days and 3 h, respectively (Fig. 2).

Analysis of the expression pattern of Smad2/3 expression in human CSKs at passage 5 treated with transforming growth factor (TGF)-β1 and amnion stromal extract (ASE) fractions. (A) TGF-β1 (10 ng/ml) only; (B) TGF-β1 and unfractionated ASE (F1); (C) TGF-β1 and ASE ≥3 kDa (F2); (D) TGF-β1 and ASE ≥30 kDa (F3); (E) TGF-β1 and ASE ≤3 kDa (F5); (F) TGF-β1 and ASE ≤30 kDa (F6); (G) ERI supplement, and (H) keratocyte basal medium (KBM) only. After treatment for 3 days, the cells were fixed and immunolabeled for Smad2/3 and F-actin using phalloidin-FITC conjugate. (I) Quantification of cells showing nuclear localization of Smad2/3 after treatments for 3 days and 3 h, respectively. The percentages of cells with nuclear Smad2/3 expression were presented as median and IQR from 10 fields of duplicated experiments. *p < 0.05 when compared to treatment with TGF-β only (Kruskal-Wallis test with Dunn-Bonferroni correction). Scale bar: 50 μm.
The fibroblast-associated F-actin fiber expression was revealed by phalloidin staining. While TGF-β1-treated cells exhibited F-actin fibers running transversely across the cells (stress pattern), the addition of various ASE fractions (except F5) with TGF-β1 resulted in predominant cortical F-actin staining (nonstress pattern) (Fig. 2). Cells treated with TGF-β1 and F5 showed transverse-aligned F-actin fibers (Fig. 2E) similar to cells with TGF-β1 only (Fig. 2A). This indicated that ASE proteins ≤3 kDa might not be beneficial to maintain CSK. In subsequent experiments, we used ASE ≤3 kDa proteins (equivalent to F2) for CSK culture.
The protein profile of F2 (proteins ≥3 kDa) was further characterized by the nano LC-MS/MS analysis. A total of 335 proteins were successfully mapped with pep tide homology ≥95% from the database of 178,828 proteins (Supplementary Table 2; https://www.seri.com.sg/wp-content/uploads/2015/07/Supplementary-Materials_Dr-Gary-Yam.pdf). We validated the candidate protein list by choosing to measure TIMP1 level using ELISA, and the concentration was 6.4 ± 4.7 ng per μg protein. Furthermore, significant pathway analysis by MetaCore™ predicted that these proteins could participate in 12 major pathways (p < 10−4 and false discovery rate <10−3). They included TGF-β signaling, cytoskeleton, and ECM remodeling, protein folding, and maturation as well as immune responses (Table 2).
Significant Pathways Potentially Affected by Amnion Stromal Extract (ASE) Proteins Using MetaCore Analysis

Adherent Culture of Primary CSK
When human CSKs at passage 3 to 5 were cultured in KBM with 0.5% FBS on collagen I-coated culture surface (104 cells/cm2) for 48 h, a number of cells exhibited stress F-actin fiber pattern (32.8%; 29.8-37.3%) (median; IQR) (Fig. 3A-I). This stress index was significantly reduced when the culture was added with ROCK inhibitor Y27632 (10 μM) (6.4%; 5.4-9.5%) (adjusted p = 0.023, Kruskal–Wallis test with Dunn–Bonferroni correction) (Fig. 3C, I). Addition of ASE F2 fraction also showed a dose-dependent reduction of stress index (6.3%; 5.1–11.1% for ASE at 0.5 μg protein/ml and 3.6%; 3–4.2% for ASE at 5 μg protein/ml) (adjusted p = 0.004 and 0.0001, when compared to cells in 0.5% FBS only) (Fig. 3I). Without Y27632, cells cultivated in 0.5% FBS and ASE (5 μg protein/ml) for 48 h had slightly higher stress index (13.2%; 12.8–20%). The stress index in serum culture was not much affected after the addition of recombinant human IGF1 (10 ng/ml) (27%; 23.3–33.3%). When CSKs were cultured with Y27632, IGF1, and ASE (5 μg protein/ml) (collectively as ERI supplement), they showed very low to negligible levels of stress index (0%, 0–3.7% with 0.5% FBS, and 0% in serum-free condition). Nevertheless, CSK marker lumican was not detectable in cells under serum culture, irrespective of the added supplements (Fig. 3B, D, F). Its expression was higher in cells cultured with serum-free ERI (Fig. 3H).

Human CSKs in various culture conditions for 48 h. (A–H) Immunofluorescence of lumican and F-actin fiber expression using phalloidin-AlexaFluor 543 conjugate in cells treated with (A, B) 0.5% FBS; (C, D) 0.5% FBS and Y27632 (10 μM); (E, F) 0.5% FBS and ERI (termed as 0.5%SERI); and (G, H) ERI supplement. Nuclei were stained with DAPI. (I) Quantification of cells showing F-actin stress fiber pattern (noncortical) after treatment with ASE, Y27632, IGF1, or their combinations under 0.5% FBS or serum-free conditions. Data are presented as median and IQR. *p < 0.05 (compared to cells in 0.5% FBS, Kruskal–Wallis test with Dunn–Bonferroni correction); **p < 0.05 (compared between cells with 5 μg/ml ASE with or without Y27632). (J–M) Human CSKs cultivated on collagen I-coated surface with (J) 0.5%SERI; (K) ERI without serum; (L) 0.5% FBS; and (M) 2% FBS. Scale bars: 20 μm.
Our result indicated that ERI suppressed human CSKs transiting to fibroblasts, even in the presence of serum. With ERI and 0.5% FBS (termed as 0.5%SERI), human CSKs appeared dendritic in morphology with cell processes extending toward the neighboring cells (Fig. 3J). The cell proliferation rate assayed by counting the mitotic figures (mitotic index) was 2.4 ± 1.4% (mean ± SD). Serum-free ERI culture generated quiescent CSKs (Fig. 3K). They had convoluted cell body with long and slender dendritic processes forming network among cells. They strongly expressed lumican and ALDH1A1 (Fig. 3H) but were devoid of stress F-actin fiber pattern (Fig. 2G). They had negligible mitotic figures (0%). Without ERI, serum-cultured human CSKs appeared as short slender shape (Fig. 3L, M). At day 6, CSKs cultured with 2% FBS had 2.4 ± 1.6% mitotic cells and those with 0.5% FBS had 0.6 ± 0.5% mitotic cells. At day 20, the mean mitotic indices were raised to 8% and 4.1%, respectively (n = 4).
Suspension Culture of Human Keratocytes
We used the hanging drop culture to study human CSKs in an adhesion-free condition. After 96 h of culture, phase contrast images were taken at the center of each drop with the focus on the meniscus level (air–liquid interface) (Fig. 4A–C). Under ERI culture, a number of cells displayed extended morphology (11.7%; 11.2–16.9%) (median; IQR), which was significantly higher than those in KBM only (0.5%; 0–1.02%) (adjusted p < 0.001) or KBM with 0.5%SERI (0%; 0–1.52%) (adjusted p = 0.01, Kruskal–Wallis test with Dunn–Bonferroni correction) (Fig. 4Di). In contrast, the formation of cell aggregates were observed in 0.5%SERI (13.6%; 12.8–14.4%) (Fig. 4C), and this was significantly reduced in culture with KBM only (0%) (adjusted p < 0.001) or with ERI (0.6%; 0–1.2%) (adjusted p = 0.018) (Fig. 4Dii).
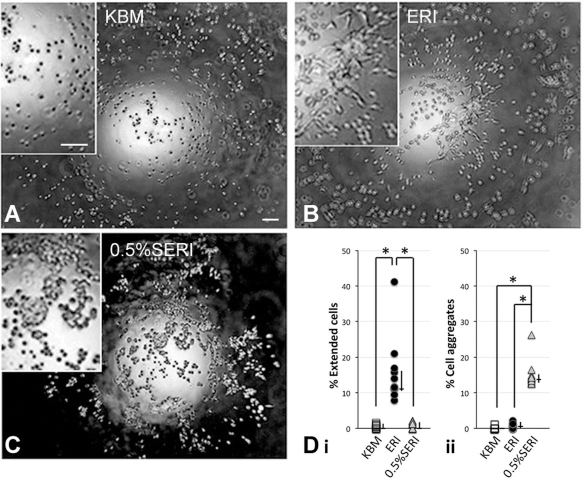
Hanging drop culture of human CSKs under (A) KBM only, (B) KBM with ERI, and (C) KBM with 0.5%SERI. Phase contrast images were captured at the meniscus level of the medium drop after 4 days of culture. (D) Percentage of cells displaying stromal-like extending sheet morphology (Di) and aggregates (Dii) were quantified from at least 10 drops, and the percentages were shown. Median and IQR are displayed next to the symbols. *p < 0.05 (Kruskal–Wallis test with Dunn–Bonferroni correction). Scale bars: 100 μm.
ERI Inhibited Fibroblast-Mediated Collagen Gel Contraction
The functional contractile activity of fibroblasts derived from human CSKs (at passage 6) was studied by the collagen gel contraction assay. For cells in KBM only, the resultant gel contraction was 40.2% (median) (IQR: 27.4– 50.1%) (Fig. 5A). It was increased to 76.1% and 74.4% for cells in KBM with 0.5% and 2% FBS, respectively (both had p < 0.05, Mann–Whitney U-test; compared to KBM only) (Fig. 5C, E, G). When TGF-β1 (20 ng/ml) was added, the median percentage of gel contraction was further raised to 91.7% (IQR: 90.2–92.4%) (Fig. 5F, G). The application of ERI almost negated this change, and the gel contraction was 36.5% (12.9–44.9%) for ERI and 42.1% (35.3–49.5%) for 0.5%SERI, respectively. Both had no significant difference when compared to cells in KBM only. Comparing CSK cultures with 0.5% FBS, there was significant reduction of gel contraction when ERI supplement was added (p = 0.002, Mann–Whitney U-test) (Fig. 5B, D, G). However, multiple comparison using Kruskal– Wallis test and Dunn–Bonferroni correction did not show any significance between these groups. Expression of cellular aSMA was observed in the contracted gels with 0.5% and 2% FBS added or not with TGF-β1 (Fig. 5C, E, F). Negligible staining was found in gels with KBM only, and KBM with ERI or 0.5%SERI (Fig. 5A, B, D).
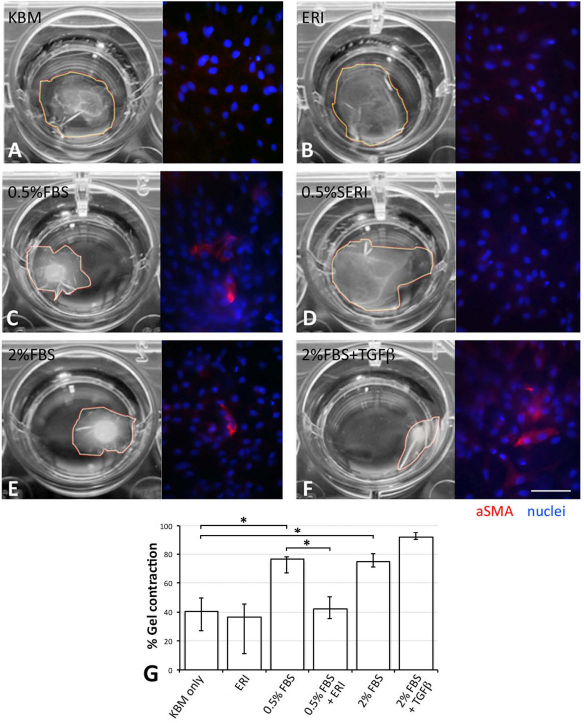
Collagen gel contractibility. Human CSKs at passage 6 were seeded to collagen gel (2.5 mg/ml) under conditions (A) KBM only, (B) ERI, (C) 0.5% FBS, (D) 0.5%SERI, (E) 2% FBS, and (F) 2% FBS with TGF-β1 (20 ng/ml). After 48 h, the resulting gel area was quantified by ImageJ software and percentage of gel area to original well area was represented as median and IQR in (G). *p < 0.05 (Mann–Whitney U-test) between indicated groups. However, multiple comparison using Kruskal–Wallis test and Dunn–Bonferroni correction did not result in any significance. Immunofluorescence of aSMA in gel was shown. Nuclei were stained with DAPI. Scale bar: 50 μm.
Proliferation of “Activated Keratocytes” and Reversion to Keratocytes
We characterized the proliferative keratocytes under 0.5%SERI culture for 6 and 14 days. By immunofluorescence, the expression of ALDH1A1, lumican, and keratocan was much weaker in cells under 0.5%SERI (Fig. 6B, F) than cells in serum-free ERI culture (Fig. 6A, E). Quantitative PCR also showed significant downregulation of ALDH1A1, lumican, and keratocan, as well as ALDH3A1 and COL8A2 in cells under 0.5%SERI culture, when compared to those under ERI condition (p < 0.05, Kruskal–Wallis test with Dunn–Bonferroni correction) (Figs. 7A and 8A). In regard to the expression levels in human corneal stromal tissue, these keratocyte markers had greater suppression in CSKs culturing with 0.5%SERI than under serum-free ERI condition. On the other hand, the cells had decreased Thy1 expression when compared to stromal fibroblasts under serum culture (Fig. 7B). This expression pattern, together with the dendritic morphology and mitotic index (2.4 ± 1.4%, as shown earlier), suggested that the “activated keratocyte” population could be expanded with 0.5%SERI.

Keratocyte marker expression in reverted CSKs from “activated keratocytes.” (A–H) Representative immunofluorescence pictures of human CSKs cultured in ERI for (A) 6 days and (E) 14 days, in 0.5%SERI for (B) 6 days and (F) 14 days, and (C) in 0.5%SERI for 3 days, followed by ERI for 3 days (mΔ) and (G) in 0.5%SERI for 7 days, followed by ERI for 7 days. (D and H) Human CSKs in 0.5% FBS for 6 and 14 days, respectively. The cells were immunolabeled for lumican, F-actin by phalloidin-AlexaFluor 543, ALDH1A1, keratocan, and aSMA. Nuclei were stained by DAPI. Scale bar: 50 μm.
We further investigated if these “activated keratocytes” could be reverted to original keratocytes and reexpress specific keratocyte genes. Human CSKs at passage 5 were cultured in 0.5%SERI for 3 and 7 days, respectively, followed by a replacement with serum-free ERI medium for another 3 and 7 days (Fig. 1). Controls were cells in either ERI or 0.5%SERI for the whole course of culture. All keratocyte genes that were suppressed in 0.5%SERI had increased expression after medium switch to ERI. By immunofluorescence, ALDH1A1, lumican, and keratocan were reexpressed at the proper subcellular locations as cells under ERI culture (Fig. 6C, G). These gene expressions were confirmed by qPCR, which additionally showed the results of ALDH3A1 and COL8A2 (Figs. 7A and 8A). Compared to 0.5%SERI culture, the gene expression after medium switch was regained (p < 0.05, Kruskal–Wallis test with Dunn–Bonferroni correction), and the recovery efficiency was more than 50%. Specifically, when examining the 14-day treatment, ALDH1A1 was recovered by 64%, ALDH3A1 by 72%, lumican by 98%, keratocan by 56%, and COL8A2 by 118%. Concomitantly, keratocan biosynthesizing enzymes, B3GNT7 and CHST6, exhibited similar pattern of changes with a reduction at 0.5%SERI culture and reexpression after switching back to serum-free ERI condition (Fig. 8A). Morphologically, the reverted keratocytes had typical dendritic shape (Fig. 6C, G).
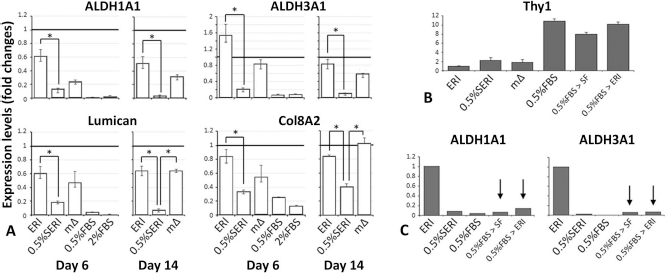
Keratocyte and fibroblast marker expression in reverted human CSKs from “activated keratocytes.” (A) Expression of ALDH1A1, ALDH3A1, lumican, and COL8A2 in human CSKs at passage 5 cultivated in ERI or 0.5% SERI for 6 and 14 days, respectively, as well as media switch (mD). Cells under serum (0.5% and 2%, respectively) culture served as controls. The gene expression level in human cornea stromal tissue was represented as a bold horizontal line. Data from quadruplicate experiments are presented as median and IQR. *p < 0.05 (Kruskal–Wallis test with Dunn–Bonferroni correction). (B) Thy1 expression in human CSKs under various culture conditions. Thy1 was generally expressed in serum-cultured cells but not in ERI cells. (C) Suppressed ALDH1A1 and ALDH3A1 expression in human CSKs cultured under 0.5%SERI and FBS-added conditions, when compared to ERI. When human CSKs cultured in 0.5% FBS for the first 3 days were changed to serum-free KBM or ERI medium, ALDH1A1 and ALDH3A1 did not reexpress (arrows).
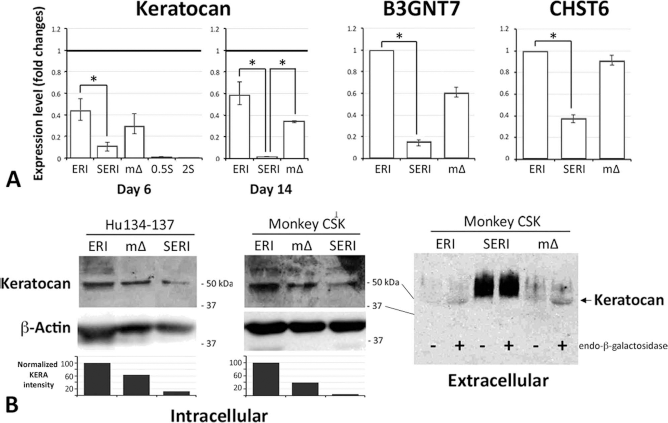
Keratocan biosynthesis, expression, and secretion. (A) Quantitative PCR analysis of the expression of keratocan and its biosynthesizing enzymes B3GNT7 and CHST6 in human CSKs at passage 5 under ERI, 0.5%SERI, and media switch (mΔ) conditions. Cells under serum (0.5% and 2%, respectively) culture served as controls. Keratocan expression in human cornea stromal tissue is represented as a bold horizontal line for the comparison with CSKs in culture. Data from quadruplicate experiments are presented as median and IQR. *p < 0.05 (Kruskal–Wallis test with Dunn–Bonferroni correction). (B) Western blot analysis of intracellular and extracellular keratocan expression in human and monkey CSKs under similar treatment as (A). The intracellular protein samples were digested with endo-β-galactosidase before SDS-PAGE and Western blotting for keratocan. β-Actin was used as housekeeping control. Band densitometry showed the changes of keratocan expression normalized with β-actin. Extracellular keratocan in monkey CSKs under similar treatment as (A) were detected by immunoprecipitation-Western blot analysis. Paired conditioned medium samples digested with (+) or without (-) endo-β-galactosidase were screened for keratocan expression.
The fibroblast marker gene Thy1 was not induced by ERI, but highly expressed in serum-containing culture, irrespective of the subsequent change of medium with or without ERI (Fig. 7B). The cells did not express keratocyte markers nor revert to keratocytes (Figs. 6D, 6H, 7C). When human CSKs cultured in 0.5% FBS for the first 3 days were changed to serum-free KBM or ERI medium, ALDH1A1 and ALDH3A1 did not reexpress (Fig. 7C, arrows).
Keratocan Expression and Secretion in Expanded CSK After Media Switch
To demonstrate the functional phenotype of CSKs, we studied the keratocan protein expression in expanded CSKs at passage 5 under ERI culture or with media switch. We detected keratocan migrating as a 50-kDa band in CSK lysates after digestion with endo-β-galactosidase (Fig. 8B). Both human and monkey CSKs had stronger expression when cultured under ERI while faint detection in 0.5%SERI for 14 days. When we performed media switch from 0.5%SERI to ERI at day 7, keratocan expression reappeared and was consistent with our qPCR finding (Fig. 8A). Band densitometry assay showed that the keratocan expression in human CSKs under 0.5%SERI was about 12% when compared to cells under ERI culture, and the level was regained to 63% after median switch. Similarly, in monkey CSKs, the keratocan expression under 0.5%SERI culture was 3.9% when compared to ERI culture and was retrieved to 39.5% after media switch (Fig. 8B). Because keratocan expression was strongly observed in the extracellular stromal matrix of in vivo corneas, conditioned media from monkey CSK cultures were also examined. The result showed that the digested samples of conditioned media from 5 × 105 cells under ERI culture for 14 days and from the media switch condition (7 days in 0.5%SERI followed by 7 days in ERI) had the 50-kDa protein band consistent with keratocan (Fig. 8B). No keratocan was detected in cell medium under 0.5%SERI. We failed to detect keratocan secretion from human CSKs, due to the insufficient cell number. Together with B3GNT7 and CHST6 expression, this indicated that ERI supplement maintained keratocan biosynthesis, expression, and secretion in vitro.
Discussion
Our work had three major findings. First, we demonstrated for the first time a successful ex vivo propagation of “activated keratocytes” by culturing human CSKs in a novel ERI supplement containing soluble ASE, ROCK inhibitor Y27632, and IGF1, and low serum concentration. Second, bona fide CSKs could be reverted from “activated keratocytes” when serum was removed, and they expressed keratocyte molecular phenotypes. This could offer a sufficient amount of genuine CSKs for corneal tissue engineering. We also characterized the protein profile of ASE by nano LC-MS/MS and showed the potential role in various cellular pathways, including TGF-β signaling, ECM remodeling for cell adhesion, and immune response.
Several studies reported that human and mouse CSKs maintained their dendritic shape and synthesized keratocan when cultured in human amnion stromal matrix, even in the presence of serum (6). The cells did not transform into fibroblasts due to the suppression of TGF-β/Smad signaling, which downregulated aSMA and fibronectin expression (29,36,56). Likewise, a reversal of myofibroblast to fibroblast phenotype was demonstrated when amniotic membrane stromal cells were placed in the amnion stromal matrix or in culture supplemented with soluble ASE (37). Collectively, these studies suggest that amnion matrix contains factors that are physiologically important to maintain keratocytes and prevent fibroblast/ myofibroblast differentiation. However, culturing cells in intact amnion stroma has limitations. The semitransparent stroma interferes with cellular visualization that is important for monitoring cell growth and contamination. The resident amniotic stromal cells, though they are sparse and damaged by deep-freezing, could also affect keratocyte adhesion and be a source of contaminants. Other issues include the need for collagenase to digest amnion stroma for CSK subpassaging and the limited scale of cell expansion. We cultured human CSKs using soluble ASE in a conventional cell culture condition, since ASE contains a complex and undefined protein profile, and some proteins could modify or be inhibitory to our goal of fibroblast suppression. We assessed different ASE fractions separated according to molecular sizes for the effect on TGF-β-associated Smad2/3 activation on human CSKs. We found that ASE proteins <3 kDa reduced this efficacy; hence the cells could transit to fibroblasts. Conversely, ASE fractions lacking <3 kDa proteins were effective in suppressing TGF-β activity, hence less fibroblast transformation. We characterized the protein profile of this ASE F2 fraction (>3 kDa) by mass spectrometry. Over 330 proteins were identified through peptide homology mapping (≥95%) using ProteinPilot analysis (out of 178,828 proteins in the database) (Supplementary Table 2; https://www.seri.com.sg/wp-content/uploads/2015/07/Supplementary-Materials_Dr-Gary-Yam.pdf). In order to validate the candidate protein list, we assayed TIMP1 expression by ELISA and detected 6.4 ± 4.7 ng TIMP1 per μg ASE protein. The positive TIMP1 expression suggested that our extraction protocol did not affect the protein profile, as TIMP1 (~23 kDa) was reported as a major protein in AM stroma (31). Further more, 12 significant pathways (p < 10−4) were annotated using MetaCore™ analysis (Table 1). Though it is not at the top significance, TGF-β-dependent induction of epithelial–mesenchymal transition was identified, validating the experimental result of ASE on TGF-β signaling and fibroblast development. Other pathways, such as cytoskeleton remodeling, cell adhesion, and ECM, as well as immune-modulatory pathways associated with anti-inflammation, antiapoptosis, and prosurvival effects were predicted. In addition, our work showed a dose-dependent antifibrosis effect on human CSKs when Y27632 was added. There were fewer cells displaying F-actin stress fiber pattern when cultured with 5 μg ASE protein/ml than those with 0.5 μg protein/ml. Therefore, ASE with its antifibrosis, growth, and survival-promoting activities, was beneficial for CSK cultivation.
Y27632 is a selective inhibitor for Rho-associated coiled-coil-containing protein kinase (ROCK) isoform (ROCK I and II). The kinase phosphorylates myosin light chain to regulate actin/myosin interaction, hence regulating the assembly and organization of actomyosin filaments in fibroblasts and myofibroblasts. In addition, it has been shown to assist cell adhesion, motility, growth, and survival in various cell types (32,45,53,58). Topical application of Y27632 to a rabbit corneal wound model resulted in an altered wound healing response by the deposition of embryonic type collagen fibrils (62). It suppressed corneal epithelial cell and keratocyte fibrosis in vitro (3,63). Human CSKs are arrested in G0 phase of cell cycle and show little regenerative capacity in vivo (cell turnover takes usually 2 to 3 years) (16). Such restricted proliferation renders ex vivo propagation of these cells difficult. Also, the highly differentiated cell character makes them poor candidates for cell culture expansion. Our work showed that low serum culture with ERI cocktail containing Y27632 at 10 mM promoted cell adhesion, survival, and proliferation. Likewise, there was significantly less F-actin stress fiber formation and collagen gel contraction. This could be attributed to the antifibrotic effect of Y27632. IGF1 is present in the corneal stroma to maintain the native CSK phenotypes, including negligible to low levels of fibroblast-associated aSMA, fibronectin, and SPARC, and high levels of lumican and keratocan expression (42). When present in collagen gel culture, IGF preserved the dendritic morphology of CSK with little collagen displacement during cell spreading, possibly due to the upregulated synthesis of ECM proteins (35). In this work, we demonstrated that human CSKs could be propagated ex vivo as “activated keratocytes” in low-serum ERI condition. They had upregulated cell survival, proliferation, and migration, without developing fibrosis and generating contractile forces.
Reversal of “activated keratocytes” to quiescent CSKs expressing the correct keratocyte profile is also a crucial requisite for translational use. By switching to serum-free ERI culture, the cells regained the keratocyte molecular pattern, including ALDH1A1, ALDH3A1, keratocan, lumican, and COL8A2. The recovery efficiencies of these genes were generally higher than 50%, compared to cells under continuous ERI culture. Specifically, at 14-day treatment, lumican expression was restored by 98%, ALDH1A1 by 64%, and keratocan by 56%. This was much higher than that reported with rabbit keratocytes in 3D collagen culture, which showed partial recovery of ALDH1A1 protein expression by 6% (55). In addition, the recovered expression of ALDH3A1, lumican, and Col8A2 were closed to 50% of human stromal tissue in vivo. While expressions of ALDH and lumican are also found in corneal epithelial cells and stromal fibroblasts, keratocan is a specific marker for stromal keratocytes. Previous reports have shown positive keratocan expression by human and mouse CSK when cultured in amnion stromal matrix, but not on plastic surface (7,29). Spheroid culture of mouse and bovine CSK with ascorbate-2-phosphate also promoted the secretion of keratan sulfate proteoglycans, including keratocan (12,64). Our work demonstrated that the media switch in the presence of ERI revived the keratocan biosynthesis (through the enzymes B3GNT7 and CHST6), RNA, and protein expression as well as extracellular secretion. This is the first time description of ex vivo expanded human CSKs could express keratocan and other keratocyte genes under a conventional 2D condition.
Our culture illustrated that the CSK yield and propagation efficiency could be affected by various factors. The amount of viable primary CSKs from stromal tissue is highly related to the donor age, cause of death, and cornea storage condition. Patel et al. reported an average CSK density in human stroma as 20,000 cells/mm3 in a group of subjects with mean age of 46 years (48). The number of CSKs decreased with age (subject group from 12 to 80 years old) at a rate of about 0.45% per year. This age-related decline was also shown in mice (44). Furthermore, CSK preservation in Optisol-GS was reported to be suboptimal. The cells underwent apoptosis, which was more significant after 5 days of storage in Optisol-GS (2). Debridement of corneal epithelium during storage further boosted CSK apoptosis. In our work, we used research human corneas preserved from 6 to 13 days in Optisol-GS, and the donor age ranged from 15 to 65 years old. Such variation of stromal tissue quality greatly influenced the stroma freshness and yield of viable primary CSKs as well as their subsequent attachment and propagation efficiency. Usually we could obtain several thousands to less than 5 × 104 viable primary cells immediately after enzymatic digestion of each corneal stroma. Inevitably, a certain quantity of them could not attach to culture surface and failed to initiate replication due to the differentiated status and cellular damages. Hence, this undermines the correct calculation of efficiency and efficacy of ex vivo CSK expansion. Instead, we showed the cell proliferation index measured by the percentage of cells exhibiting mitotic figures to be about 2.4% under 0.5%SERI culture. As an average, this could expand human CSKs for up to six to eight passages (some samples could be expanded for 10 passages) while maintaining the keratocyte phenotype and the cell number could reach about 5 × 106 to 107 per human cornea. As each human cornea is estimated to have about 106 keratocytes (based on the measurement of corneal diameter ~11.5 mm, thickness of 0.5~0.6 mm, stroma spanning ~90% of corneal volume, and reported CSK density ~20,000 cells/mm3 stroma), this indicates that our propagation protocol could generate CSKs sufficient for making 5 to 10 full-thickness artificial stroma for transplantation use.
Appropriate tissue processing skill is also critical for primary CSK viability and yield. Trimming into tiny stromal fragments would ease the collagenase digestion efficiency to release the resident CSKs. This procedure is preferably conducted by clean vertical cuts instead of slicing action in order to minimize cellular damages. As shown by different morphological studies of human stromal tissue, the stellate-shaped CSKs align along the direction of collagen fibrils and form extensive network with numerous diffuse and thread-like cell processes making contacts with adjacent keratocyte processes (27,52). This poses the vulnerability of cells when exposed to shear or tangential stress. In addition, overdigestion with collagenase should be avoided to maintain cell viability and its good adhesive ability in primary culture.
Taking these factors into consideration, our refined protocol greatly improved the efficiency of propagating CSKs. We have not proven whether the expanding cells are CSSCs or progenitor cells. However, as the primary cells were harvested from central stromal button rather than peripheral region, we believe that the differentiated CSKs are expanded via the stage of “activated keratocytes.” Our work on ex vivo keratocyte propagation, together with recent reports of the generation of keratocyte-like cells from hCSSCs or hESCs and their experimental injection to defective stroma to restore corneal phenotype and transparency (4), indicate the plausibility of cell-based therapy for corneal disorders. It may offer an alternative to the invasive keratoplasty for the treatment of corneal stromal disorders.
Footnotes
Acknowledgments
This work was supported by Biomedical Research Council Translational Clinical Research Partnership Grant (TCR0101673), Singapore. We also thank the Department of Gynecology of Singapore General Hospital, SingHealth, and the Singapore Eye Bank, Ministry of Health for human amnion collection. We appreciate the Confocal Microscopy Laboratory of National Cancer Center, SingHealth for confocal microscopy. The authors declare no conflicts of interest.