
Abstract
Impaired peripheral wound healing is a hallmark of diabetis pathology and has been attributed to compromised macrophage activation. Stroke is another component of diabetic pathology, with increased tissue infarction and worsened recovery although the mechanisms remain unresolved. In this study, we investigated whether a compromised glial/macrophage response might contribute to cerebral hypoxic-ischemic (H/I) brain damage in diabetic (db/db), relative to their normoglycemic db/+ mice. Hypoxia-ischemia was induced in 8-week-old male db/db and db/+ mice by the ligation of right common carotid artery followed by systemic hypoxia (8% O2: 92% N2) for 17 mins. Mice were killed at specific intervals of reperfusion/recovery and the brains analyzed by in situ hybridization or total RNA isolation. In situ hybridization using bfl-1 (microglia) and glial fibrillary acidic protein (GFAP) (astrocytes) revealed expression of both bfl-1 and GFAP in the ipsilateral hemisphere at 4 h in the db/+ mice, which was delayed and minimal in the db/db mice. RNase protection assays showed a robust increase in expression of the proinflamatory cytokines tumor necrosis factor-α (TNFα), interleukin-1 IL-1α, and IL-1β mRNA in the db/+ mice at 6 to 8 h of reperfusion peaking at 8 to 12 h; in db/db mice expression was markedly delayed and diminished. Real-time-polymerase chain reaction (RT-PCR) confirmed the reduced and delayed expression TNFα, IL-1α, IL-1β, and the growth factors insulin-like growth factor-1 and ciliary neurotrophic factor in the db/db mice; enzyme-linked immunosorbent assays confirmed the reduced and delayed translation of IL-1β protein. These findings suggest that a compromised inflammatory response may underlie the greater infarct associated with diabetic db/db mice compared with their nondiabetic littermates following a hypoxic/ischemic insult.
Introduction
Impaired wound healing is a significant complication in Type I and Type II diabetics. Wounds fail to heal quickly and healing is inhibited by both extrinsic factors such as neuropathy, macro- and microvascular diseases, and intrinsic factors such as altered bioavailability of growth factors and abnormalities of extracellular matrix and protease activity (Loots et al, 1998). A variety of in vitro and in vivo studies have indicated that the macrophages that secrete the growth factors and cytokines that mediate the inflammatory response and promote wound healing after an injurious stimuli, are disrupted in diabetic patients (Clinton and Libby, 1992). Moreover, direct application of growth factors and cytokines has been successful in promoting the healing of ulcers and wounds in both diabetic animals and human subjects (Greenhalgh et al, 1990; Wheeler and Brodie, 1998).
Another complication of Type I and Type II diabetes is cerebral ischemia, or stroke. Diabetic patients are at an increased risk for stroke, with increased mortality and morbidity after a stroke (Bonow and Gheorghiade, 2004). Experimental animal models of stroke have confirmed that the extent of tissue damage is significantly worse in diabetic animals, although the mechanistic basis for this remains unknown. We have previously shown that female diabetic db/db mice fail to exhibit an early microglial (brain macrophage) inflammatory response and had greater tissue damage as compared with age-matched female nondiabetic db/ + mice (Zhang et al, 2004). Thus, we hypothesized that an impaired inflammatory response in the brain contributes to the enhanced damage after stroke in the diabetic animal. To test this hypothesis in the present study, we investigated the expression of the cytokines and growth factors, which contribute to the early immune response of the brain. Employing an experimental stroke model of unilateral carotid artery ligation and systemic hypoxia—ischemia (H/I) (Vannucci et al, 2001) we compared the time course and extent of cytokine release in the ischemic and nonischemic hemispheres in male db/db and db/+ mice. We observed a significantly delayed increase in the expression of the proinflammatory cytokines (tumor necrosis factor-α (TNFα), interleukin-1 (IL-1α, and IL-1β) responsible for initiating microglial and astrocytic activation, and a decrease in the expression of the growth factors ciliary neurotrophic factor (CNTF) and insulin-like growth factor-1 (IGF-1) after stroke in the db/db mice compared with db/+ controls.
Materials and methods
Animals
Male C57BL/KsJ db/db mice and nondiabetic littermates (db/+) were purchased from Jackson Laboratories (Bar Harbor, ME, USA) at 7 weeks of age. At 8 weeks of age, animals were subjected to unilateral cerebral H/I (8% O2/balance N2, 17 mins, 35.5°C) as previously described (Vannucci et al, 2001). Briefly, animals were anesthetized with isoflurane (4% in 70% N2O:30% O2), and a small incision was made in the neck. The right carotid artery was exposed and double-ligated with 4-O surgical silk, the incision was sutured, and the animals allowed to recover with access to food and water for 3 h. Systemic hypoxia was induced by exposure to 8% O2/balance N2 in temperature-controlled glass chambers (35.5°C). At the end of the 17-mins hypoxic interval, animals were allowed to recover in room air, and returned to their cages with free access to food and water, supplemented with 5% (w/v) glucose. The combination of unilateral carotid artery ligation with systemic hypoxia produces ischemia (50% to 60% reduction in normal cerebral blood flow (CBF) in the hemisphere ipsilateral to the ligation (Vannucci et al, 2001). As CBF is restored to normal on return to normoxic conditions this is a model of transient unilateral cerebral ischemia, which results in reproducible brain damage in the ipsilateral, but not the contralateral, hemisphere (Vannucci et al, 2001).
At 4, 6, 8, 12, 24, and 48 h of recovery from H/I (i.e., reperfusion after return to normoxia) the mice were killed, the brains were removed and divided into contralateral and ipsilateral hemispheres and stored in RNA later solution (Ambion Inc., Austin, TX, USA) for RNA isolation and analysis.
In addition, brains from comparably treated animals were removed and frozen in isopentane (–40°C). Cryosections (16 μm) were cut for in situ hybridization studies. All animal protocols were approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee.
RNase Protection Assays
Total RNA was isolated from tissue using TRI reagent (Invitrogen Life technology, Molecular Research Center Inc., Cincinnati, OH, USA), according to the manufacturer's instructions. RNase protection assays (RPA) were performed using a customized mouse template, which includes macrophage inhibitory factor (MIF), interferon-γ (IFNγ), macrophage colony stimulating factor (MCSF), IL-18, IL-12p40, IL-6, IL-10, IL-1α, IL-1β, TNFα RNAs (Riboquant custom, PharMingen, San Diego, CA, USA). Probes labeled with 32P-UTP were hybridized with extracted RNA according to the manufacturer's instructions. Purified samples were separated using a 6% ureapolyacrylamide gel electrophoresis sequencing gel and the fragments were visualized by phosphorimage analysis and quantitated using Image Quant software (Molecular Dynamics, Sunnyvale, CA, USA). Gene expression was normalized to the respective levels of glyceraldehyde-3-phosphate dehydrogenase mRNA.
Real-Time Polymerase Chain Reaction
Total RNA was isolated from ipsilateral and contralateral hemispheres as described above. 2 μg RNA was used to generate cDNA template using the Omniscript RT kit by Qiagen according to the manufacturer's protocols. Sample of cDNA template (2 μl) was treated with labeled primer 1 and unlabeled primer 2 for TNFα, IL-1α, IL-1β, CNTF, and 18S as internal control (Lux Fluorogenic Primer, Invitrogen Life Technology) at a final concentration of 10 μmol/L. Platinum Quantitative PCR Super mix—UDG with ROX reference reagent (Invitrogen Life Technology, CA, USA) was used as described by the manufacturer. For IGF-1 quantitation, 2 μl cDNA template was treated with FAM™ dye-labeled TaqMan® MGB probe and mouse-specific IGF-1 primer and 18S as internal control (TaqMan Primer, Applied Biosystems) at a final concentration of 250 nmol/L for the probe and 900 nmol/L for each primer. TaqMan Universal PCR master mix (Applied Biosystems CA, USA) was used as per the manufacturer's instruction. The reaction plate was placed in Applied Biosystems 7300 Real Time PCR System, programmed for the real-time-polymerase chain reaction (RT-PCR). Preliminary experiments were performed to validate the primers and to determine optimal annealing temperature and cycling parameters. Data were analyzed using sequence detection system 1.9.1 software (Applied Biosystem, CA, USA).
In situ Hybridization
Cryosections (16 μm) were analyzed for IL-1β, bfl-1, and GFAP mRNA expression by in situ hybridization. A 500 bp mouse IL-1β plasmid was sequenced to confirm their identity and transcribed by using T7 and SP6 polymerase for antisense and sense, respectively. It was linearized by enzyme Xba1 for antisense and HindIII for sense.
A 663-bp mouse bfl-1 cDNA probe was cloned by PCR from a mouse kidney library by using primers 5′-TTCCAA CAGCCTCCAGATATGATTAGGG and 5′-GTGGGAGCCAA GGTTCTCTCTGGTCCG It was sequenced and transcribed by enzyme SP6 and T7 and linearized by Apa1 and Spe1 for antisense and sense, respectively. Similarly 1159 bp of rat GFAP was transcribed by T7 and SP6 polymerase for antisense and sense, respectively and linearized by PVU II for both sense and antisense. These probes were used to generate 35S-labeled riboprobes for in situ hybridization as previously described (Vannucci et al, 1997; Zhang et al, 2004) with slight modifications of hybridization temperature and salt treatment. Hybridization of IL-1β and bfl-1 was performed at 65°C (50%, SSC/formamide) in a humid chamber box for 16 h. To enhance specificity slides were treated with 0.05 × SSC at 60°C for 30 mins. For GFAP, hybridization was performed at 58°C and washed with 0.1 × SSC at 50°C for 15 mins. After the initial autoradiographic exposure, all sections were dipped in Kodak NTB3 photographic emulsion, developed, and counter stained with hematoxylin.
Enzyme-linked Immunosorbent Assay
Contralateral and ipsilateral hemispheres of db/+ and db/db mice were collected at 8, 12, 24, and 48 h after H/I, frozen on dry ice, and stored at −80°C. Tissues were homogenized in a 2-[4-(2-hydroxyethyl)-1-piperazinyl] ethanesulfonic acid, ethylenediamine-N,N,N′,N′-tetraacetic acid, sucrose (20:1:255 mmol/L) buffer using a Ten-Broeck ground glass homogenizer. Homogenates were centrifuged at 50,000g (Beckman Coulter Optima™ Max Ultracentrifuge) for 20 mins. The supernatants were assayed for the presence of IL-6 and IL-1β by using cytokine-specific capture/detection antibody pairs (Ray Biotech Inc., GA, USA); recombinant cytokines were used as standards. Experiments were performed according to the manufacturer's instruction (Protein concentration of each sample was measured by using BCA™ protein assay kit (PIERCE, Rockford, IL, USA).
Statistical Analysis
Data were analyzed by one-way analysis of variance followed by Tukey analysis for multiple comparisons with Graph Pad Software, Prism 2.01. Relative expression software tool (REST®) was used for the statistical analysis of RT-PCR data, significance was assumed at P < 0.05.
Results
Hypoxia—Ischemia in Male db/db and db/+ Mice
Our previous study suggesting a microglial dysfunction after H/I in the diabetic animal was conducted in female db/db and db/+ mice (Zhang et al, 2004). Therefore, it was important to show that the same phenomenon occurs in the male. Male db/db and db/+ mice were subjected to 17 mins of H/I, and the early expression of bfl-1 (microglia) and GFAP (activated astrocytes) mRNA was analyzed by in situ hybridization (Figure 1). Bfl-1 expression was apparent as early as 4 h of recovery in both the striatum and adjacent cortex of the db/+ mice, but was barely detectable even at 8 or 12 h of recovery in the db/db mice and the intense striatal expression of bfl-1 observed in the normoglycemic db/+ was not detected in the diabetic mice at any time of recovery. These patterns of bfl-1 expression persisted for 3 to 5 days in both groups of animals, as was previously shown for the females (data not shown). Figure 1 also illustrates that astrocytic activation as evidenced by enhanced GFAP expression was diminished and delayed in the diabetic mice. As with bfl-1, the increased GFAP mRNA expression was apparent at 4 h in the ipsilateral hemisphere of the db/+ animals, with heightened expression in the striatum over the subsequent 24 h. GFAP-positive cells appear to ‘surround' the region of bfl-1-positive microglia, as has been reported before in nondiabetic mouse brain (O'Donnell et al, 2002). In the diabetic mice the expression of GFAP mRNA was barely detectible at 4 h and significantly decreased throughout the ipsilateral hemisphere, indicative of tissue infarction. Thus, both the increase in tissue damage, as well as the reduced microglial response previously reported for the female db/db mouse is characteristic of the H/I response of the male diabetic brain as well.
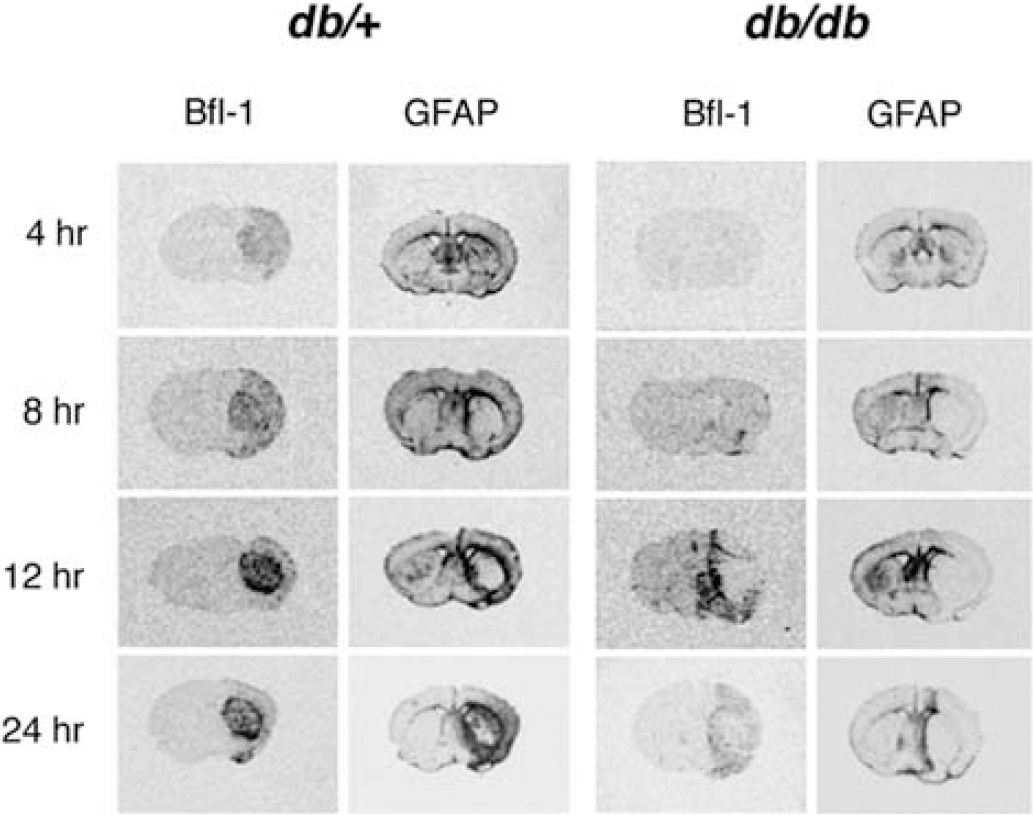
Temporal activation of microglia and astrocytes in db/+ and db/db mice after H/I. Bfl-1 and GFAP in situ hybridization. Time course of bfl-1 and GFAP mRNA expression in db/+ and db/db brains after 17 mins of H/I. Cryosections (16 μm) were analyzed by in situ hybridization using 35S-labeled riboprobes as described in ‘Materials and methods’. Shown are representative autoradiograms of coronal sections for db/+ and db/db mouse brains at the level of striatum.
Microglial and astrocytic activation after an injury represents the brain's wound healing response and, as in the periphery, is mediated by the autocrine and paracrine actions of released cytokines. To investigate our hypothesis that wound healing is impaired after H/I in the diabetic brain, we determined whether the apparent delay in microglial activation in the db/db mice was associated with decreased cytokine expression. Total RNA was isolated from ipsilateral and contralateral hemispheres of db/db and db/+ mice at different times after H/I and the levels of expression of the cytokines MIF, IFNγ, MCSF, IL-18, IL-12p40, IL-6, IL-10, IL-1α, IL-1β, and TNFα were analyzed with an RPA.
Figure 2 depicts the ratio of mRNA levels of the cytokines TNFα, IL-1α, IL-1β, IL-6, MIF, and MCSF in paired ipsilateral and contralateral hemispheres during reperfusion. Values were first normalized for recovery with the house keeping gene glyceraldehyde-3-phosphate dehydrogenase. No differences in the levels of gene expression between db/+ and db/db mice were observed before H/I (data not shown). The first changes in expression of TNFα, IL-1α, and IL-1β were detected in the ipsilateral hemisphere of the db/+ at 6 h; levels further increased at 8 and 12 h of recovery. In the db/db mice levels of expression were barely detectable until 12 h of recovery but by 24 h were not different from the nondiabetic controls. By contrast, IL-6 expression did not increase until 8 h and no differences in expression were observed between nondiabetic and diabetic animals. MIF and MCSF expression levels were elevated in the ipsilateral hemispheres of both the db/+ and db/db at 4 h, and remained so through 24 h. Similar to IL-6, there were no significant differences in MIF expression between the db/+ and db/db mice, whereas MCSF expression was significantly reduced in the db/db mice at 6 and 8 h of recovery compared with their nondiabetic controls. With the exception of MIF expression, which remained elevated, all other mRNA levels returned to baseline levels by 48 h of recovery.
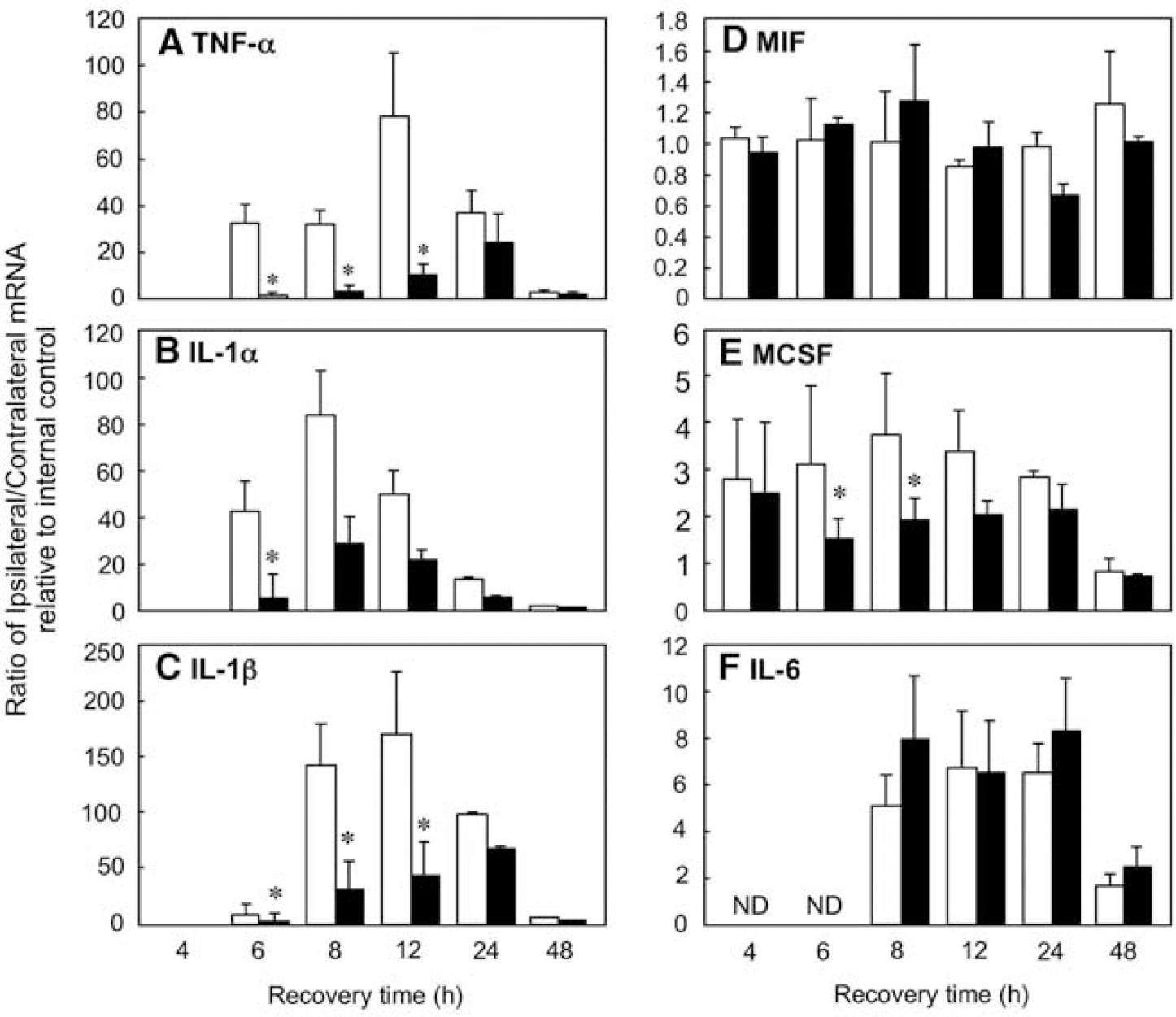
RPA of proinflamatory cytokine expression in db/+ and db/db mice after H/I: time course. Total RNA isolated from ipsilateral and contralateral hemispheres of db/+ and db/db mouse brains was analyzed by RPA for TNFα (
To confirm the results obtained from RPA analysis, RT-PCR was performed with primers for the proinflammatory cytokines TNFα, IL-1α, and IL-1β, and the growth factors CNTF and IGF-1. The results are again expressed as a ratio of paired ipsilateral to contralateral hemispheres, relative to their respective internal controls (Figure 3). Real-time-polymerase chain reaction analysis of RNA isolated at various times post-H/I revealed increased TNFα expression in the ipsilateral hemisphere of the db/+ mice as early as 4 h, which remained six- to tenfold elevated through 24 h of recovery. By contrast, TNFα expression, although detectable at 4 h in the db/db mice, was only increased three- to five-fold the first 12 h and remained reduced relative to the db/+ mice throughout recovery. Consistent with RPA analysis, small but significant increases in the levels of IL-1α and IL-1β were apparent at 6 h of recovery in db/+ mice relative to the db/db mice. At 8 h of recovery, the levels of both IL-1α and IL-1β increased 30-fold in the ipsilateral hemisphere of the db/+ mice compared with less than five-fold changes observed in the db/db mice. Expression of both cytokines in the db/db mice approached nondiabetic levels at 12 and 24 h of recovery. Consistent with the RPA data, all cytokine expression in the ipsilateral hemisphere of both groups of animals returned to baseline levels by 48 h. It should be stressed that the changes in expression of the respective cytokines determined by either RPA or RT-PCR are corrected for recovery of house keeping genes and are independent of absolute mRNA levels.
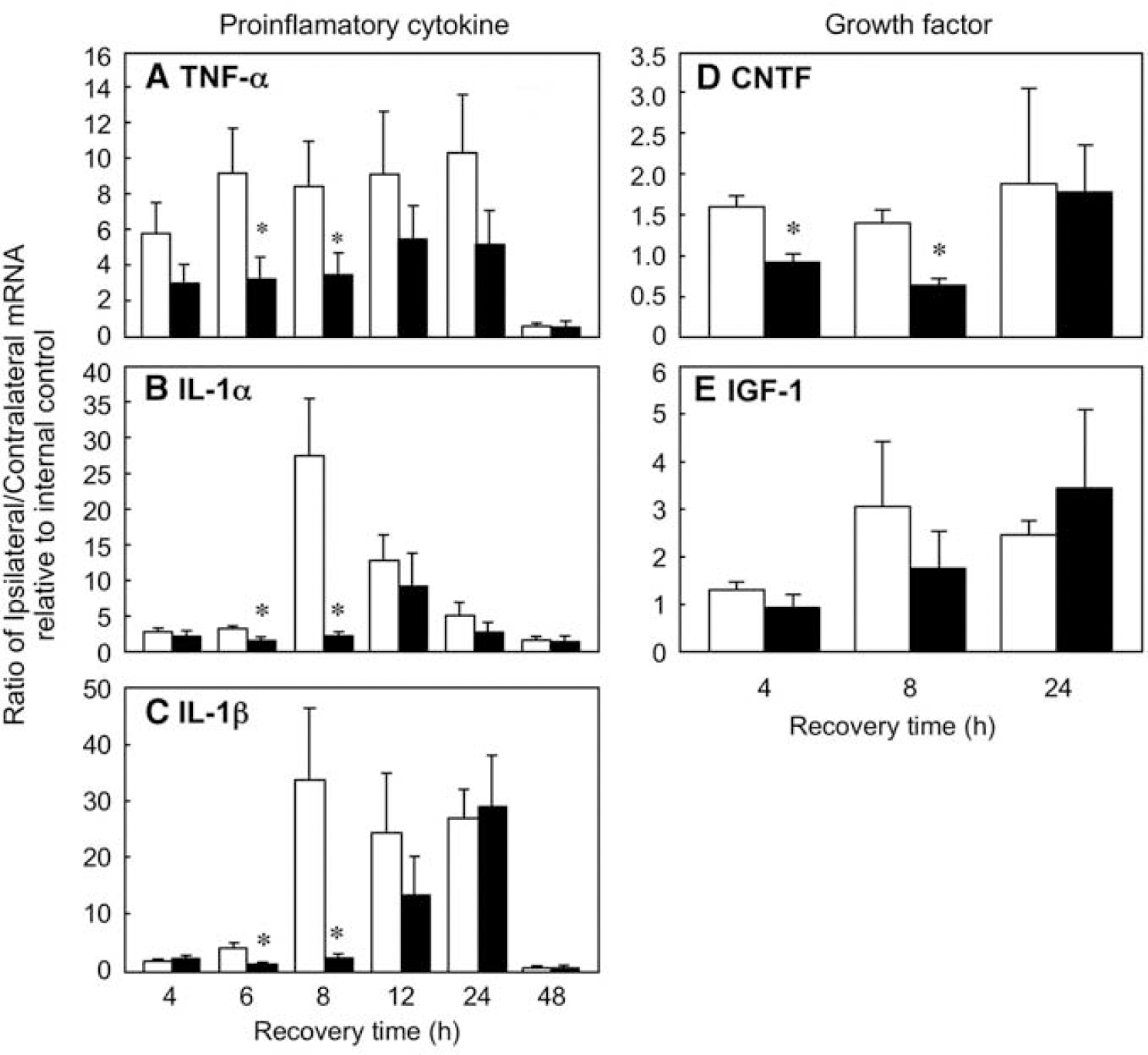
Real-time-polymerase chain reaction analysis of proinflamatory cytokine and growth factor expression in db/+ and db/db mice after H/I: time course. Total RNA isolated from ipsilateral and contralateral hemispheres of db/+ and db/db mouse brains was analyzed by RT-PCR for TNFα (
Another important component of reactive gliosis is the release of growth factors that promote proliferation and cell survival. Two such growth factors are IGF-1, which is expressed primarily in microglia in response to H/I (O'Donnell et al, 2002), and CNTF, which is predominantly astrocytic in origin. To investigate the effect of diabetes on the expression of these growth factors after H/I, RNA was isolated from ipsilateral and contralateral hemispheres of db/+ & db/db mice at 4, 8, and 24 h recovery from H/I. Modest but significant increases in CNTF expression were seen early in recovery in the db/+ animals that were not observed in the db/ db mice (Figure 3). A similar pattern of IGF-1 expression was observed, however, the differences in expression between control and diabetic mice did not reach statistical significance.
While RT-PCR provides a methodology to quantitate changes in mRNA expression, it offers no information on the pattern of expression or cellular localization relative to the evolution of the infarct. IL-1β expression was determined by in situ hybridization at the level of the striatum and anterior hippocampus at various times of recovery and representative autoradiograms are presented in Figure 4. As anticipated, the pattern of IL-1β expression in both the db/+ and db/db mice parallels that of bfl-1 illustrated in Figure 1, both temporally and spatially, thus supporting a predominantly microglial localization for the increased IL-1β expression.
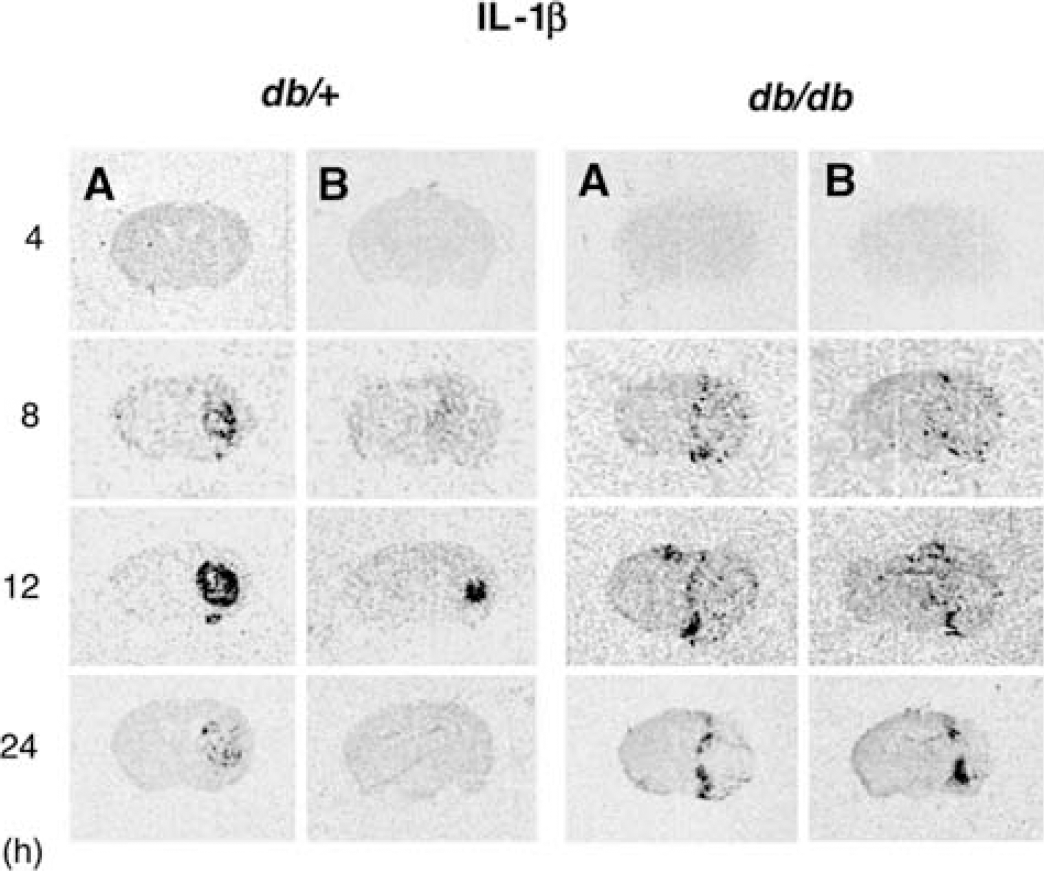
Time course of IL-1β expression in db/+ and db/db after H/I: In situ hybridization. Time course of IL-1β mRNA expression in db/+ and db/db brains after 17 mins of H/I. Cryosections (16 μm) were analyzed by in situ hybridization using an 35S-labeled riboprobe as described in ‘Materials and methods’. Shown are representative autoradiograms of coronal sections for db/+ and db/db mouse brains at the level of striatum (
To determine whether the changes in IL-1β mRNA observed above were translated into protein, a separate cohort of animals were subjected to H/I and ipsilateral and contralateral hemispheres were isolated and homogenized as described in Materials and methods. The homogenates underwent centrifugation at 50,000g for 20 mins and the resultant supernatants were analyzed by an enzyme-linked immunosorbent assay (ELISA) for the presence of IL-1β and IL-6. Figure 5 illustrates the respective levels of IL-1β and IL-6 in the ipsilateral hemispheres of animals at various time points during recovery. Cytokine levels in contralateral hemispheres were minimally elevated in the db/+, but not db/db brain. A biphasic pattern of IL-1β levels was observed in the ipsilateral hemisphere of the db/+ animals. At 8 h of recovery the levels of IL-1β were markedly elevated in the ipsilateral hemisphere of the db/+ mice and these levels appear to decline at 12 h of recovery before rebounding at 24 h and ultimately declining at 48 h of recovery. By contrast, in the ipsilateral hemisphere of the db/db animals, the peak of IL-1β protein is not observed until 12 h of recovery and expression remains elevated in some but not all animals at 24 h of recovery before receding towards baseline levels at 48 h. These data are in marked contrast to that observed with IL-6. In the ipsilateral hemisphere of the db/+ mice the levels of IL-6 remained low at 8 and 12 h of recovery but were significantly increased at 24 h before returning to baseline levels at 48 h. In the db/db, the levels IL-6 were higher than in the db/+ at all times of recovery and also peaked at 24 h. However, unlike in the db/+ mice they also remained elevated at 48 h of recovery.
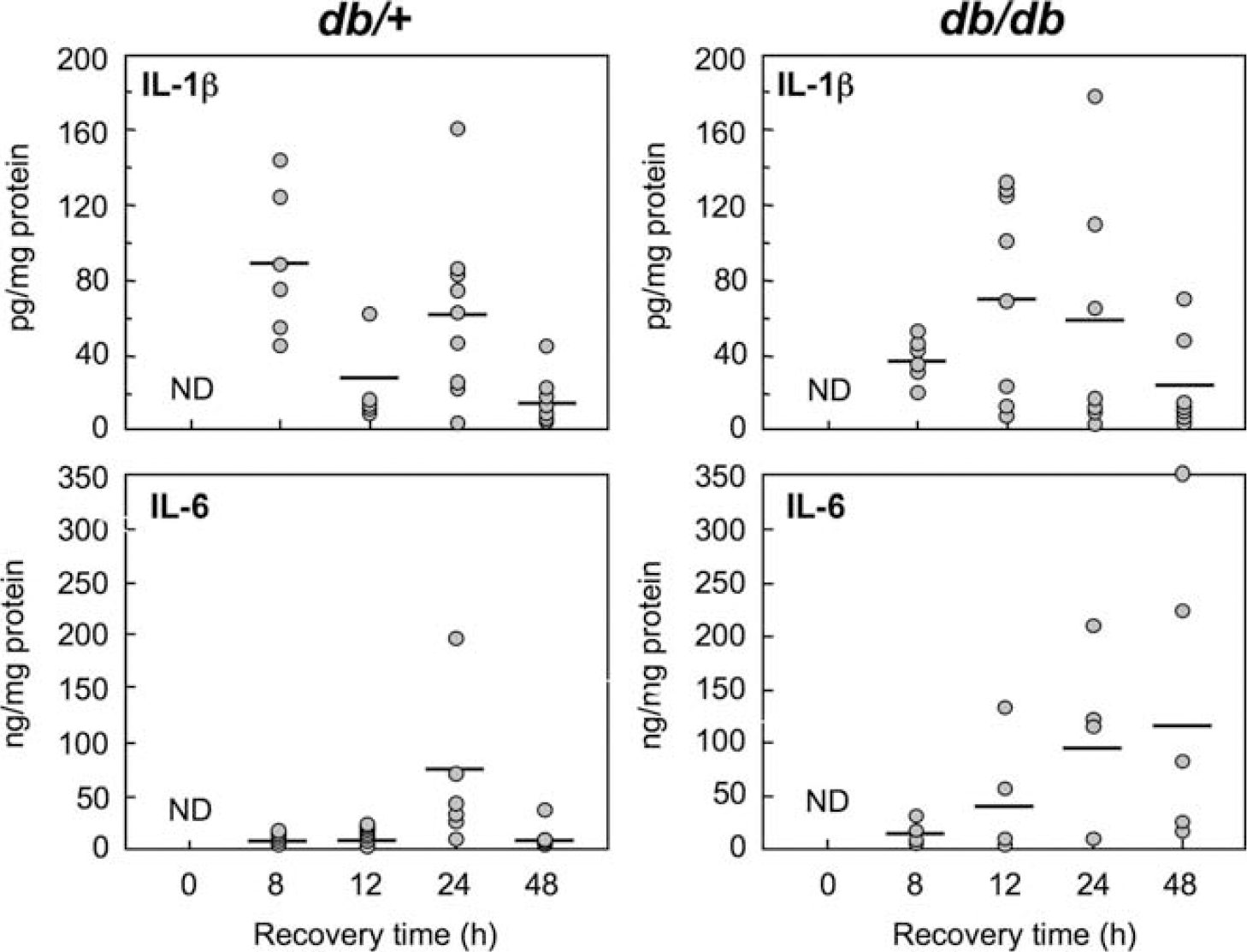
IL-1β and IL-6 protein levels in ipsilateral hemispheres of db/+ and db/db mice at various times after H/I: ELISA analysis. Homogenates of ipsilateral hemispheres of db/+ and db/db mice were centrifuged at @ 50,000g for 20 mins and resultant supernatant analyzed by ELISA for IL-1β and IL-6 at various times after 17 mins H/I, as describe in ‘Materials and methods’. Data expressed as scatter diagram with means denoted by—as either pg (IL-1β) or ng (IL-6) per mg of supernatant protein. (n = 5 to 9). ND—nondetectable.
Discussion
It is well established that diabetic patients have a higher incidence of stroke coupled with greater mortality and morbidity during recovery than their nondiabetic counterparts (Bonow and Gheorghiade, 2004; Mankovsky and Ziegler, 2004). The increased cell death after stroke observed in the diabetic patients and in several different rodent models of diabetes has been consistently attributed to the excess accumulation of lactic acid, acidosis, and reduced recovery of affected tissue (Folbergrova et al, 1992; Siesjo, 1988). However, studies by Nedergaard and Diemer (1987) and Nedergaard et al (1988) showed that preischemic hyperglycemia associated with diabetes elicited greater tissue loss after MCAO than in control animals made equivalently hyperglycemic, suggesting factors other than simple lactoacidosis are involved. This hypothesis was further supported by our own studies in db/db mice, in which we found higher levels of circulating glucose and longer durations of lactoacidosis in diabetic female db/db mice than their male counterparts despite finding the volume of the infarct was substantially smaller in the female mice (Vannucci et al, 2001). In a subsequent study in female db/db mice, we observed that microglial activation after a H/I insult was both diminished and delayed when compared with the their db/+ nondiabetic littermates (Zhang et al, 2004).
In the current study in male db/+ and db/db mice, we confirm the delay in microglial activation in the male db/db mice compared with their nondiabetic littermates after H/I (Figure 1). Microglial activation was monitored by following the mRNA expression of the antiapoptotic protein bfl-1, which, is an excellent microglial/ macrophage marker. In Figure 1 we also show a comparable delay in the expression of GFAP in ipsilateral hemisphere of the db/db mouse after H/I, suggesting a delay in astrocytic activation.
Microglial and astrocytic activation, and the initiation of the inflammatory response after H/I, are essentially autocrine/paracrine activities. The initiation is mediated by the cytokines IL-1β and TNFα that are released in response to an initial triggering event such as local ischemia by the microglia. In the current study, we have investigated the time course for the increase in expression of a variety of cytokines in the ipsilateral hemispheres of male db/db and db/+ mice during recovery from H/I. Using RPA, we have clearly showed a delay in the onset of expression of both IL-1β and TNFα as well as other proinflammatory cytokines and growth factors in the ipsilateral hemisphere of the db/db mice after H/I and we subsequently confirmed these observations by RT-PCR. The early expression of TNFα, IL-1α, IL-1β, IL-6, and MCSF, as well as the growth factors CNTF and IGF-1, were observed in the ipsilateral hemisphere of db/+ mouse by 4 to 6 h post-H/I, peaking at 8 to 12 h before returning to base line by 48 h. By contrast, the increase in the expression of the proinflammatory cytokines TNFα and IL-1β was delayed until 12 h of recovery in the db/db mouse and peaked at 24 h of recovery before also returning to base line levels by 48 h.
The temporal cytokine responses observed here in the control db/+ mice are entirely comparable to those seen in previous studies of stroke and other brain trauma. It has been shown that IL-1β is produced within 15 mins of cortical injury by activated microglia (Herx et al, 2000; Herx and Yong, 2001) and its expression was essential for subsequent astrocyte activation and the expression of CNTF. The induction of TNF mRNA has been reported to peak at 5 h and return to normal levels by 24 h in a perinatal rat model of cerebral H/I (Szaflarski et al, 1995). In a rat model of transient focal ischemia expression of TNFα and IL-1β mRNA was detected as early 1 h and peaked at 3 and 6 h, respectively (Wang et al, 1995). The induction of TNFα and IL-1β have been shown to be induced early in other brain injuries, for example, in a stab-wound injury model in adult mouse brain, elevation of IL-1β and TNFα mRNA were observed at 6 to 12 h after the injury (Rostworowski et al, 1997).
The measurement of changes in cytokine mRNA expression provides early indicators of the activation of the inflammatory response, however, ultimately it is their translation that is required for the response to be sustained. We were able to measure the temporal changes in IL-1β and IL-6 by ELISA. We observed significantly greater levels of IL-1β in the ipsilateral hemisphere of db/+ mouse compared with db/db after 8 h after H/I. In the db/+ mice the concentration of IL-1β declined at 12 h before peaking by 24 h whereas in diabetic mice, the initial increase appeared to be delayed until 12 h but again returned towards baseline levels at 48 h. The levels of IL-6 also peaked at 24 h post-H/I in both db/+ and the db/db. A similar biphasic response of IL-1β has been observed in the gerbil hippocampus after transient global ischemia (Saito et al, 1996) where elevated levels of the proteins were seen at 3 to 6 h after occlusion, which declined sharply at 12 h before transiently increasing at 24 h and then returning to base line by 48 h. However, in a rat permanent MCAO model, the initial peak of IL-1β was observed at 4h and then a progressive increase over subsequent 3 days of recovery (Legos et al, 2000). In both of these stroke models the IL-6 levels remained elevated at 48 to 96 h after occlusion. In our earlier study (Zhang et al, 2004), we showed a profound increase in the number of macrophage present in the ipsilateral hemisphere of the female db/db mice at 48 h of recovery, which we were able to confirm in the males db/db (Kumari et al, 2005, data not shown). The elevated levels of macrophages could well account for the increases in the concentration of IL-6 that is observed in the ipsilateral hemisphere of the db/db mice.
The parallels between the compromised inflammatory responses in the db/db mice observed in this study and the numerous diabetic peripheral wound-healing studies are striking. Disturbances in the inflammatory and proliferation phases of wound healing in the diabetic patient have been shown to delay all phases of recovery from angiogenesis to maturation/remodeling (Greenhalgh, 2003; Jeffcoate et al, 2004). Several wound healing studies in rodent models of diabetes, including the db/db mouse, have shown that the release of cytokines and growth factors from macrophages and their phagocytic activity are compromised (Bitar and Labbad, 1996; Cooper et al, 2001; Doxey et al, 1998; Fantuzzi and Faggioni, 2000; Greenhalgh, 2003; Zykova et al, 2000). Another characteristic of the compromised peripheral wound healing response in both the diabetic mouse and patient is the decreased proliferating potential and migration of fibroblasts Goldstein et al, 1979; Lerman et al, 2003), a situation that appears to parallel the delayed and impaired astrocytic response observed in this study.
Delays in microglial activation after injury have also been shown in mice that are unable to respond to IL-1β owing to a lack of expression of the gene encoding IL-1β or the IL-1 type I receptor (Basu et al, 2005; Herx et al, 2000). As mentioned above, cilliary neurotrophic factor (CNTF) expression is greatly enhanced in astrocytes after injury and together with IL-1 promotes astrocyte nuclear hypertrophy (Albrecht et al, 2002; Dallner et al, 2002; Hudgins and Levison, 1998). In this study, we observed a significant upregulation CNTF mRNA at 4 and 8 h of recovery in the ipsilateral hemisphere of nondiabetic mice but no corresponding increase in CNTF mRNA was observed in the diabetic mice until 24 h of recovery, consistent with the delay in the onset of expression of the proinflammatory cytokines TNFα and IL-1β.
Considerable controversy surrounds the extent to which the proinflammatory responses elicited by TNFα and IL-1β play a positive or negative effect in recovery from stroke (for reviews see Lucas et al (2006), Schwartz (2003), and Streit (2002)). Clearly reactive gliosis and the formation of a gliotic scar are essential for the re-establishment of the patentcy of the CNS. However, in animals where IL-1β or its receptor have been knocked out or where IL-1β has been neutralized by antibodies or endogenously added receptors, there have been clear demonstrations of improved outcome after stroke, suggesting a deleterious role for promotion of the inflammatory response. In the case of TNFα, both deleterious and neuroprotective activity have been ascribed to the cytokine (Hallenbeck, 2002) in the recovery from stroke.
This study further supports the Janus face the inflammatory response plays in recovery from injury. In this study, we report that in diabetic mice after a stroke there is delayed and diminished cytokine release that is in turn reflected in delayed and reduced microglial and astrocytic activation and ultimately greater cell loss. Thus, interventions that reduced the inflammatory activation would not be predicated. These responses clearly mimic those seen in peripheral wound healing where impairments in the diabetic wounds are attributed to aberrations in macrophage responsiveness. Some success in reversing this situation has been achieved by the application of growth factors such as PDGF, FGF, TGF-β either separately or in combination both systemically and topically (for reviews see Greenhalgh (2003) and Jeffcoate et al (2004)).