
Abstract
Genetic alterations have recently been described as emerging during the culture of embryonic stem cells or induced pluripotent stem cells, raising concerns about their safety in future clinical use. Myoblasts are adult stem cells with important therapeutic potential that have been used in clinical trials for almost 20 years, but their genome integrity has not yet been established. Here we produced 10 human myoblast preparations and investigated their genomic stability. At the third passage, half of the preparations had a normal karyotype and half showed one to four alterations/30 metaphases. Chromosome 2 trisomy was found in 1–2/30 meta-phases and/or 2/100 nuclei by FISH in 3/10 samples, and there was no other recurrent anomaly. When prolonging cultures, these erratic abnormalities were never associated with a growth advantage. Cellular senescence was manifested in all samples by growth arrest before passage 15. Expression of TERT was always negative. Molecular analysis of individual p53 transcripts did not reveal tumorigenic mutations. CGH array (10 samples) and exome sequencing (one sample) failed to detect copy number variations or accumulation of mutations, respectively. Myoblasts did not grow either in soft agar or in vivo after injection in immunodeficient mice. Hence, occasional genomic abnormalities may occur during myoblast culture but are not associated with risk of transformation.
Introduction
Stem cells are endowed with considerable therapeutic potential for regenerative medicine. Nevertheless, the probability of genetic alterations emerging during the culture of these cells represents a risk for their potential clinical use. This was recently shown to be the case for embryonic stem cells (16) and induced pluripotent stem cells (4). As genetic instability is a hallmark of cancer (10), these observations have raised concerns regarding the safety of embryonic stem cell therapy products.
Potentially, adult stem cells are susceptible to chromosomal instability during their expansion in vitro. This could theoretically be the case for cells such as myoblasts that can be grown in culture from the satellite cells adjacent to muscle fibers. Indeed, their production necessitates at least 3 weeks of cell culture to reach sufficient numbers for therapy, that is, more than 108 cells. Among adult stem cells, myoblasts are endowed with important therapeutic potential for treating inherited or acquired muscle diseases. Hence, they have been used in human clinical trials for almost 20 years in an attempt to treat muscular dystrophies (11,13–15,20,21,23,24,32), heart failure (3,17), or urinary (2,22) and fecal incontinence (8). Clinical trials performed with myoblasts of either allogeneic or autologous origin have shown excellent tolerance, and cellular transformation has never been reported. However, the genomic integrity of myoblasts has not been investigated to date. Hence, we analyzed in terms of identity, purity, viability, and genomic safety profile (karyology, tumorigenicity) 10 batches of human myoblasts that were produced in agreement with the requirements of Part IV of Annex I to Directive 2001/83/ EC from the European Union. Notably, we studied the chromosomal stability of myoblast preparations and the possible consequences of instability on the risk of transformation using a similar approach to that recently reported for mesenchymal stem cells (MSCs) (29).
Materials and Methods
Production of Human Myoblasts
Muscle samples were obtained from 10 different individuals (eight females, two males; mean age ± SD; 54 ± 20.3) undergoing surgery under general anesthesia. Written informed consent was obtained from all donors. The biological collection was authorized by the French Ministry of Research after approval by the local institutional ethics committee (reference DC-2009–996). Cell preparation was adapted from a previously used myoblast preparation protocol (27) according to patent EP1572988A1. Briefly, for each donor, a muscle fragment was minced and enzymatically digested with collagenase NB4 (Serva Electrophoresis, Heidelberg, Germany). Then, isolated cells were cultured in a myoblast selection medium containing 10% fetal calf serum (HyClone, Logan, UT, USA) for approximately 3 weeks. Myoblasts were then amplified for approximately 1 week in an expansion medium containing 10% fetal calf serum and 10 ng/ml basic fibroblast growth factor (R&D Systems, Minneapolis, MN, USA). The cell therapy finished product corresponds to cells harvested at the third passage (P3). These cells were produced for the need of this study and were not injected into humans.
Cellular Immunophenotyping
Immunophenotype was performed by flow cytometry using a FC500 apparatus (Beckman Coulter, Villepinte, France) after staining 0.5–1 × 106 cells with combinations of the following fluorochrome-labeled monoclonal antibodies: cluster of differentiation 56-phycoerythrin–cyanine 5.1 (CD56-PC5, 1:100; clone B159, Beckman Coulter), CD90-fluorescein isothiocyanate (FITC) (clone 5E10, 1:40; BD, Le Pont de Claix, France), CD45-phycoerythrin (PE) (clone DW124–5-2, 1:20; Beckman Coulter), CD34-FITC (clone 581, 1:10; Beckman Coulter), and human leukocyte antigen (HLA)-I FITC (clone B9.12.1, 1:5; Beckman Coulter).
Clonogenic Assays
To evaluate the frequency of colony-forming units (CFU), P3 myoblasts were seeded at low density (20 cells/ well) into six-well plates (BD Falcon, Franklin Lakes, NJ, USA) in expansion medium. When cells within colonies reached 80% confluence (approximately 10 days), clones were stained with Giemsa (RAL diagnostics, Martillac, France), counted, and the frequency of CFU was determined as the total number of clonal cellular clusters formed divided by the number of cells initially seeded.
Myogenic Differentiation
P3 myoblasts were seeded at low density (20 cells/well) into six-well plates in expansion medium. When cells within colonies reached 80% confluence (approximately 10 days), they were further incubated at low fetal calf serum concentration (1%) for 1 week before staining with Giemsa to identify myogenic clones containing myotubes.
Growth Kinetics
Cells were cultivated at an initial density of 700–1,000 cells/cm2, and passages were performed when the surface occupied by myoblasts on the plate reached 80%, until growth arrest was observed. The doubling time (DT) was calculated as follows: DT = culture time/N, where N is the number of divisions determined as log (number of cells recovered/number of cells plated)/log2.
RT-PCR Analysis
Total RNA was extracted with TRIzol (Invitrogen, Cergy Pontoise, France). cDNA was prepared by reverse transcription of 1 μg of total RNA. Primers used were desmin, 5′-CCA ACA AGA ACA ACG ACG-3′ F and 5′-TGG TAT GGA CCT CAG AAC C-3′ R (408 bp); muscle-specific regulatory factor 4 (MRF4), 5′-GAT CCC ACC GAC CCT TCC TGG-3′ F and 5′-GAG GCT AGA CCT AAG CCA CTC GC-3′ R (215 bp); myogenic factor 5 (Myf5), 5′-CCA GGC TTA TCT ATC ATG TGC TAT G-3′ F and 5′-GTT AAG CAT TGC AAC AAG CTA CCC-3′ R (320 bp); myogenic differentiation (myoD), 5′-CGA TAT ACC AGG TGC TCT GAG GG-3′ F and 5′-GGG TGG GTT ACG GTT ACA CCT GC-3′ R (430 bp); paired box 7 (Pax7), 5′-GTA CGG CCA GAG TGA GTG CCT G-3′ F and 5′-CTG TTG GAG CCA TAG TAC GGA AG-3′ R (245 bp); telomerase reverse transcriptase (TERT), 5′-TTG TCA AGG TGG ATG TGA CG-3′ F and 5′-ATG TAC GGC TGG AGG TCT GT-3′ R (196 bp); glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′-TGC CAT CAA CGA CCC CTT CA-3′ F and 5′-TGA GCT TGC CCA CAG CCT TG-3′ R (569bp). PCR was performed using 35 cycles (94°C–60°C–72°C) except for TERT (94°C–61°C–72°C). GAPDH was used as a housekeeping gene, and a normal muscle sample was used as a positive control. A sample in which RNA had been omitted at the step of reverse transcription was used as negative control. cDNA from a human embryonic kidney (HEK-293; female; ATCC, Molsheim, France) tumor cell line was used as a positive control of the TERT RT-PCR.
Cytogenetic Analysis
Karyotype was determined on passage 3 (P3) myoblasts and also on myoblasts obtained after prolonging cultures in expansion medium (P4 and P8 cell preparations). P3, P4, and P8 cell preparations were seeded in short-term cultures so as to obtain usable metaphases for karyotyping. In parallel, a smear of these cell suspensions was made on slides and frozen for fluorescence in situ hybridization (FISH) analysis. For each karyotype, 30 metaphases (rather than 20 metaphases when performing antenatal diagnosis) were analyzed to detect eventual abnormalities in the number or structure of chromosomes. If aneuploidy was evidenced in at least one of the three passages for a given myoblast preparation, it was subsequently sought for specifically by FISH for each of the three passages (P3, P4, and P8). FISH was performed according to standard protocols and to the manufacturer's manual. Specific TelVysion subtelomeric probes (Abbott Molecular, Abbott Park, IL, USA) were used to hybridize the two telomeric ends of the chromosomes involved.
Comparative Genomic Hybridization (CGH) Array Analysis
Total DNA was extracted from 10 muscle biopsies and corresponding P3 or P4 primary myoblast cultures using the Flexigen extraction kit (Qiagen, Courtaboeuf, France). High-resolution CGH array analysis was performed using Agilent SurePrint G3 Human 4 × 180 K catalog arrays (G4449A, Agilent Technologies, Santa Clara, CA, USA). One microgram of DNA from P3 myoblasts and from its corresponding muscle samples was labeled with Cy3 or Cy5 fluorochromes, respectively, using Agilent Oligo CGH Microarray Kit as recommended by the manufacturer. Similarly, P4 DNA was compared to P3 DNA. Hybridization results were extracted with Feature Extraction Software (10.5.1.1, Agilent Technologies) and analyzed with DNA analytics software (version 4.0.81, Agilent Technologies). The data were processed using the ADM-2 algorithm, with the threshold set at 5.3 SD. A rearrangement was defined by any deviation of a log ratio greater than or less than 0.3 on at least three consecutive probes.
Exome Analysis
Genomic DNA was extracted from muscle or from myoblasts using Flexigen extraction kit (Qiagen). Exons of genomic DNA samples were captured using Agilent in-solution enrichment methodology. Sequence capture, enrichment, and elution were performed according to manufacturer's instruction and protocols (Human All exon kit V4 + UTRs, 70 Mb, Agilent) without modification. Massively parallel sequencing of paired-end 75 bases was performed on Illumina HiSEQ2000 (Illumina, San Diego, CA, USA). Briefly, 3 μg of each genomic DNA were fragmented by sonication and purified to yield fragments of 150–200 bp. Paired-end adaptor oligonucleotides from Illumina were ligated on repaired A-tailed fragments, then purified and enriched by six PCR cycles. We then hybridized 500 ng of these purified libraries in the SureSelectoligo probe capture library (Agilent Technologies) for 24 h. After hybridization, washing, and elution, the eluted fraction was PCR amplified with 10 to 12 cycles, then purified and quantified by quantitative PCR. Each eluted-enriched DNA sample was then sequenced on an IlluminaHiSEQ2000 as paired-end 75b reads. Image analysis and base calling were performed using Illumina Real Time Analysis (RTA) Pipeline version 1.14 with default parameters.
The bioinformatics analysis of sequencing data was based on the Illumina pipeline (CASAVA1.8). CASAVA performs alignment of the reads to the human reference genome (hg19) with the alignment algorithm ELANDv2 (performs multiseed and gapped alignments), then calls the single nucleotide polymorphism (SNPs) based on the allele calls and read depth, and detects variants (SNPs and Indels). Only the positions included in the bait coordinates were conserved. Genetic variation annotation was performed using IntegraGen in-house pipeline, which consists of gene annotation (RefSeq), detection of known polymorphisms (dbSNP 132, 1000 Genomes) followed by mutation characterization (exonic, intronic, silent, nonsense, etc.). For each position, exomic frequencies (Homo and HTZ) were determined from the entire IntegraGenExome database and the exome results provided by HapMap. Results were provided per sample, including coverage/ depth statistical analysis per exome and per target. A list of somatic variants in the coding regions and three intronic bases corresponding to consensus splicing sites was generated. Common polymorphisms were removed by comparison to dbSNP131, 1000 Genomes Project data, and the IntegraGen in-house database comprising 50 exomes. Quality control filtering removed variants sequenced in less than five reads, with QPHRED of <10 and with sequence size not included within the range represented by the mean size ± 3 SD. Using the Integrated Genomics Viewer (IGV)32, we assigned muscle genome as reference genome and compared samples of myoblasts P3 and P8 to the reference.
p53 Functional Assay
Functional assays in yeast for tumor protein p53 (TP53) mutation detection was performed as previously described (7). Briefly, mRNA was purified from P3 myoblasts with QuickPrep Micro mRNA purification kit (GE Healthcare, Nantes, France). cDNA was synthesized with a First-Strand cDNA synthesis kit (GE Healthcare). PCR was performed with primers specific for TP53 as described (7), except that the DNA polymerase used was PrimeSTAR (Takara Bio, Inc., Shiga, Japan). Ten microliters of PCR products was directly used for yeast transformation. After incubation for 3 days at 30°C on selective medium, the number of white and red colonies was counted. More than 92% white colonies was required to conclude wild-type TP53 mutation, as previously described (7).
Culture in Soft Agar
In vitro assessment of tumorigenicity was performed by soft agar transformation assay in six-well plates. First, a 2× expansion medium containing 20% fetal calf serum and 20 ng/ml of basic fibroblast growth factor was prepared. Each well was coated with 1 ml of a 1:1 mixture of 2× expansion medium and a solution of 1.2% melted agar (Invitrogen). After solidification of this first layer, P3 myoblasts were added as 1.5 ml of a 2:1 mixture of the cell suspension (1 × 104 cells/ml) in 2× expansion medium at 37°C and the 1.2% agar solution. After solidification of this second layer, 0.5 ml of 2× expansion medium was added on top and changed once a week. After 4 weeks at 37°C in a humidified atmosphere with 5% CO2, plates were microscopically examined for colony formation. HeLa tumor cells were used in place of myoblasts as positive control.
In Vivo Tumorigenesis
Adult nonobese diabetic/severe combined immunodeficient (NOD/scid) mice (Charles River Laboratories, Chatillon-sur-Chalaronne, France; n = 10; four males, six females) were randomized to receive in the left gastrocnemius either 106 myoblasts recovered at P3 or HeLa tumor cells in 50 μl of physiologic saline. Animals were maintained in good conditions with access ad libitum to food and fresh water. Mice were monitored for tumor formation, and caliper measurement was performed in case a tumor was observed (A, difference of length compared to uninjected muscle; B, difference of width). Tumor volume was determined as AB2/2. Mice were sacrificed to weigh gastrocnemius muscle after 60 days of follow-up or when the tumor reached 1,500 mm3. Histological analysis was performed on serial sections encompassing the entire injected region. Sections were stained with anti-HLA-class I (HLA–ABC, M0736, 1:100; Dako, Glostrup, Denmark), anti-human lamin A/C (NCL-LAM-A/C, 1:50; Leica, Nanterre, France), or anti-Ki-67 (M7240, 1:100; Dako) monoclonal antibodies. Immunostainings were revealed using a peroxidase-labeled anti-mouse Ig antibody (K3954, Dako) and analyzed by an experienced pathologist unaware of the experimental groups. This protocol was approved by the regional institutional ethics committee under the reference N/03–01–12/03/01–15.
Statistical Analysis
Muscle weight data were expressed as median ± 25th percentile and compared using the Mann–Whitney test. Percentages of tumor-free mice were compared using the log-rank test. Differences were considered statistically significant when p < 0.05.
Results
Phenotypic Characterization of Myoblast Preparations
We analyzed 10 preparations of human myoblasts (referred to as M1 to M10) that were obtained from a muscle biopsy after 3–4 weeks of culture under the French Good Practices for Cell Therapy Products (Bulletin Officiel 2010/11, December 15, pages 91–140, http://www.sante.gouv.fr/fichiers/bo/2010/10–11/ste_20100011_0001_p000.pdf). These cell preparations were harvested at the third passage (P3), which corresponded to 27 ± 3 days of culture (mean ± SD). All cell preparations displayed a characteristic phenotype of myoblasts, that is, CD56+, CD90+, CD45-, CD34-, and HLA-I+ (Fig. 1A). On average, their doubling time was 30 h (range 24–43 h), 25% of seeded cells gave rise to colonies (range 13–39% CFU) (Table 1), and evidence of myogenic potential was found (Fig. 1B). All cell preparations expressed mRNA transcripts of typical molecular markers of myogenic cells such as desmin, MRF4, Myf5, MyoD, and Pax7 (Fig. 1C).

Characterization of human myoblast preparations. (A) Example of immunophenotype for myoblast preparation M3: flow cytometry analysis after staining with anti-cluster of differentiation 56 (CD56), −CD90, −CD45, −CD34, and −human leukocyte antigen (HLA)-I monoclonal antibodies. Complete phenotypic results appear in Table 1. (B) Example of myogenic differentiation. Giemsa staining of myotubes obtained after in vitro differentiation of myoblast preparation M9. (C) RT-PCR analysis of desmin and myogenic factors [muscle-specific regulatory factor 4 (MRF4), myogenic factor 5 (Myf5), myogenic differentiation (myoD), and paired box 7 (Pax7)] gene expression. cDNAs were from the 10 different myoblast preparations (M1 to M10) and from one normal muscle sample. Ctrl, negative control in which RNA was omitted from reverse transcription step.
Immunophenotype and Growth Characteristics of Myoblast Preparations

CD, cluster of differentiation; HLA, human leukocyte antigen, DT, doubling time; CFU, colony-forming unit.
Clonogenicity assay providing the frequency of seeded cells that give rise to colonies.
Transient Karyotypic Abnormalities May Occur in a Minority of Myoblasts
Karyotype analysis performed on 30 metaphases at passage P3 revealed that 5/10 myoblast preparations (M2, M4, M5, M6, and M9) had a normal karyotype (Table 2), although 2% of the nuclei of myoblast preparation M4 displayed, when analyzed by FISH, a trisomy 2, which was not found on the karyotype. For the other five myoblast preparations at P3 (M1, M3, M7, M8, and M10), abnormalities were found in 1–4/30 metaphases, although the mosaic aneuploidy in preparations M3 and M7 was not confirmed by FISH. Two different structural chromosomal alterations were found in 1/30 metaphases of preparations M1 and M10 at P3. In samples M1, M4, and M8, chromosome 2 trisomy was found in 1–2/30 metaphases and/or 2/100 nuclei by FISH. There was no other recurrent karyotypic abnormality.
Karyotypic Characteristics of Myoblasts
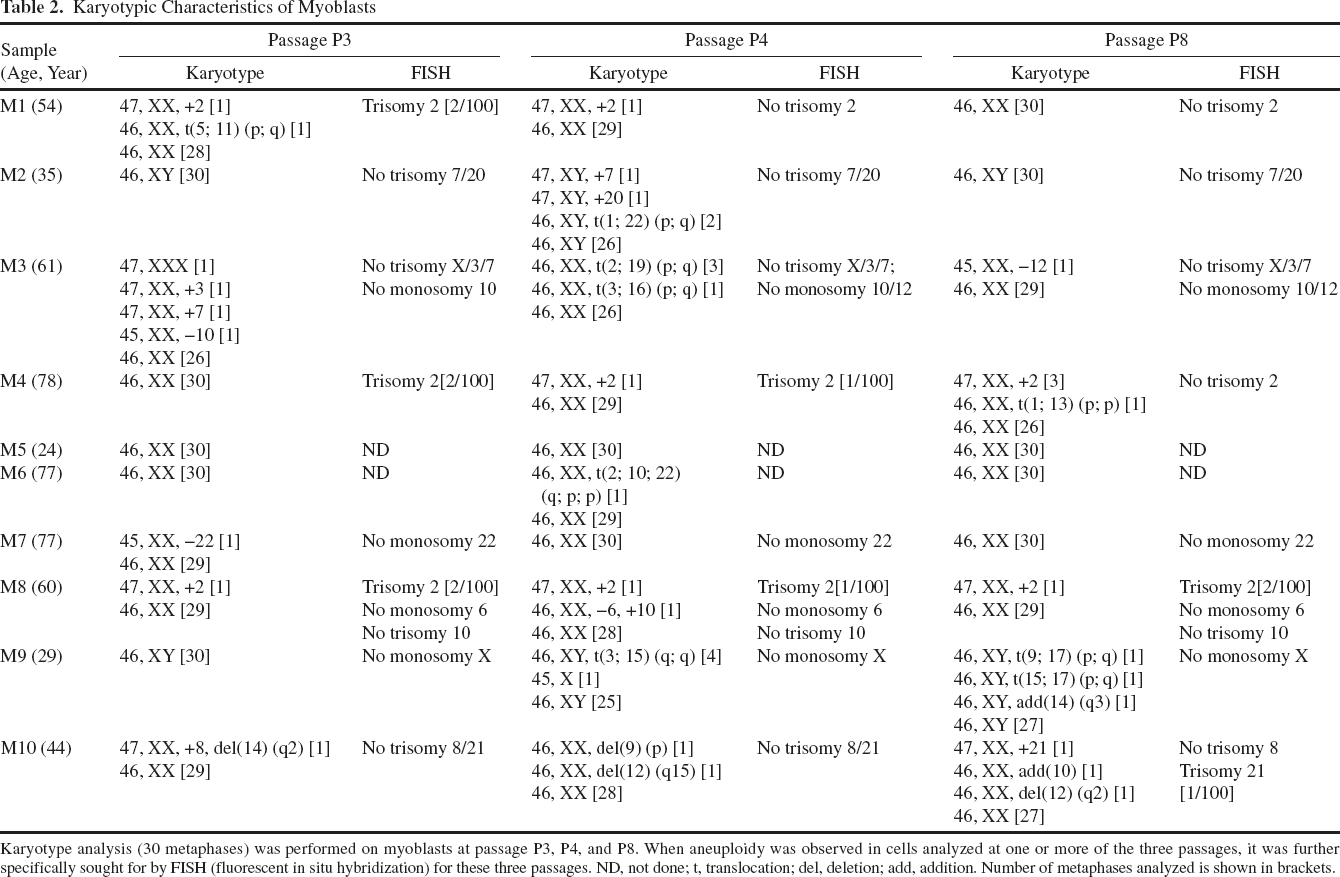
Karyotype analysis (30 metaphases) was performed on myoblasts at passage P3, P4, and P8. When aneuploidy was observed in cells analyzed at one or more of the three passages, it was further specifically sought for by FISH (fluorescent in situ hybridization) for these three passages. ND, not done; t, translocation; del, deletion; add, addition. Number of metaphases analyzed is shown in brackets.
To evaluate whether karyotypic abnormality could confer a selective advantage to the cell or rather is detected in cells that are prone to die, we prolonged culture of the P3 cell therapy preparations up to passages 4 and 8 and reiterated the karyotypic and FISH analyses. Importantly, the individual karyotypic anomalies found at P3 disappeared when prolonging cultures, except for trisomy 2 in preparation M8, which remained present in 1/30 metaphases. In myoblast preparations M2, M4, M6, and M9 that displayed a normal karyotype at P3, various chromosomal alterations could later arise at P4 or P8 in a minority of cells, underlining the erratic character of these transient anomalies.
The lack of expansion in culture and disappearance at further passages indicated an absence of selective advantage for the cells presenting chromosomal abnormalities. This was further supported by the results of a genome-wide CGH array study, which failed to detect any change in gene copy numbers when comparing DNA extracts from all 10 myoblast preparations at passage P3 to the corresponding muscle DNA from which they were derived (Fig. 2A). No copy number variation was observed either in preparation M1 to M4 when comparing passages P4 to P3 (Fig. 2B).
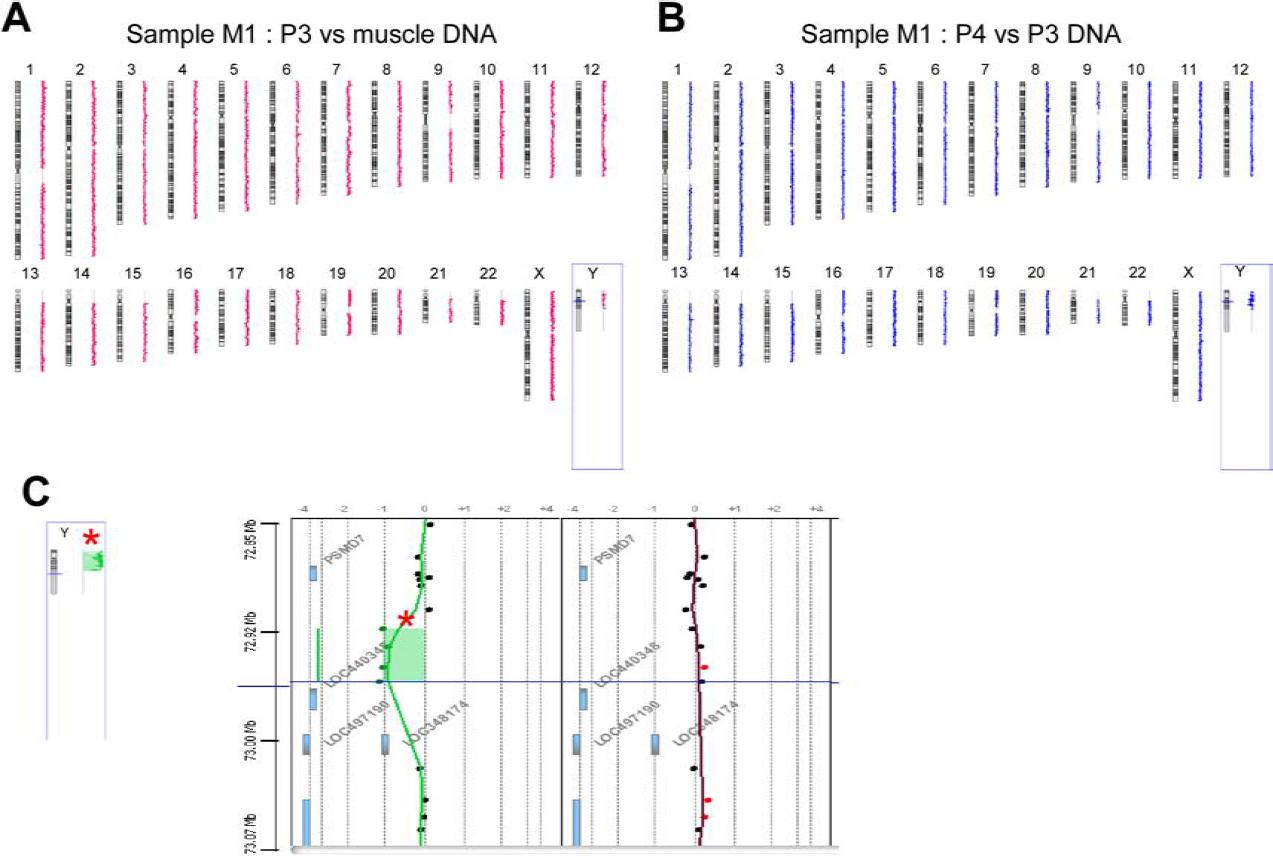
Absence of detectable variation in the number of gene copies in myoblast DNA by comparative genome hybridization (CGH) array pangenomic analysis. (A) Example of comparison of gene copy numbers between DNA of the myoblast preparation M1 at passage 3 (P3) and the original muscle sample at day 0. CGH array analysis of P3 in comparison to muscle DNA for all myoblast preparations (M1 to M10) revealed no statistically significant abnormality. (B) Example of comparison of gene copy numbers between DNA of myoblast preparation M1 at P4 and P3. CGH array analysis of P4 compared to P3 for the four myoblast preparations studied (M1 to M4) revealed no statistically significant abnormality. (C) As control, presence of significant variations (red star) in copy numbers between female (M1) and male (M2) myoblasts at P3 for the Y chromosome (left panel). Example of an autosomal polymorphic copy number variation (right panel, green dots: log ratio = −1).
Besides karyotypic changes and copy number variations, acquisition of somatic mutations that could generate a selective advantage during culture would also be a drawback for cell therapy because of the risk of oncogenesis. In order to systematically examine the possible occurrence of mutations during the culture of myoblasts, we performed exome sequencing of muscle DNA and of passages P3 and P8 DNA from the M6 myoblast preparation using next generation sequencing. We sequenced 70 Mb for the three samples, each base being covered by an average depth of 80×. The number of variants compared to a reference genome appeared similar in the different samples (not shown), reflecting genomic stability during cell culture. Using muscle tissue as genome reference, we did not find any nonsynonymous variant affecting coding regions at passages P3 and P8. In line with the above-mentioned observations, this analysis indicated an absence of acquired mutations during the cell preparation process and a lack of selective advantage for clonal populations in culture.
Myoblasts Enter Senescence After Prolonged Culture and Do Not Present Tumorigenic Potential
Cellular stability was investigated by evaluating senescence in prolonged culture, by assaying telomerase (TERT) gene expression and by analyzing individual p53 transcripts, as well as by performing in vitro and in vivo assays of tumorigenesis. We first analyzed the long-term growth capacities of myoblast preparations. All of them entered senescence, as manifested by growth arrest between passages P9 to P14, which corresponded to 47 to 88 days of culture (Fig. 3A). The maximum number of divisions, observed for M9, was 55 doublings. Whereas the cell therapy product corresponds to cells harvested at P3, lengthening culture was accompanied by a progressive loss of CD56 positivity, presumably reflecting the propensity of CD56+ myoblasts to enter senescence when culture is prolonged (Fig. 3B). In line with this lack of long-term growth capacity, there was an absence of TERT reexpression in all myoblast preparations (Fig. 3C).

Replicative senescence of human myoblasts. (A) Cumulative number of cell divisions before senescence (growth arrest) according to the number of cell passages. (B) Frequency (mean ± SEM) of CD56+ cells along passages for all myoblast preparations (M1 to M10). (C) RT-PCR analysis of telomerase reverse transcriptase (TERT) gene expression in myoblast preparations at P3. Human embryonic kidney (HEK) cells were used as a positive control.
The behavior of myoblasts in soft agar culture was determined. None of the 10 myoblast preparations could grow in culture in these conditions, in contrast to the HeLa tumor cells used as a positive control (Fig. 4A).

Lack of tumorigenicity of human myoblast preparations. (A) Culture in soft agar was performed for 4 weeks with myoblast preparations M1 to M10 at passage P3. This assay showed lack of development for all 10 myoblast preparations. One example is shown for M1 and HeLa tumor cells used as positive control. Magnification is 50×. (B) Tumor protein p53 (p53) mRNA functional assay in yeast. Example for RNA from one myoblast preparation (M7, left panel) compared to RNA extracted from a tumor where p53 is mutated as positive control (red colonies, central panel) and from a cell line with wild-type p53 as negative control (white colonies, right panel). RNA from all 10 myoblast preparations (M1 to M10) revealed as wild-type using this test. (C, D, E, F) HeLa cells (positive control; n = 5), 106, or myoblasts (n = 10) were injected intramuscularly into the left gastrocnemius of nonobese diabetic severe combined immunodeficient (NOD scid) mice. The uninjected right gastrocnemius was used as control. Percentage of tumor-free mice (C). Gastrocnemius muscle weight (median ± 25th percentile) at euthanasia at day 60 (D). Photographs of muscle at day 60 after myoblast injection or when HeLa control tumor reached 1,500 mm3 (E). Histological analysis of muscles injected with HeLa cells (left) or with M9 myoblast preparation (right) after staining with anti-HLA class I (top), anti-human lamin A/C (middle), or anti-Ki-67 (bottom) antibodies. Results show presence of calcifications around the site of injection (upper right) but lack of human cells at day 60 postinjection. Similarly, no HLA-positive, lamin-positive, or Ki-67 cells were found in any of the five other (M2, M3, M7, M8, and M10) samples analyzed (F).
We next performed molecular analysis of individual p53 transcripts in yeast using a test that allows identification of transformation-prone mutations as red colonies (7). This revealed an absence of tumorigenic mutations for all myoblast preparations, as illustrated with the example of M7 preparation (Fig. 4B).
Finally, each of the 10 cell preparations was injected intramuscularly into one NOD scid immunodeficient mouse (four males, six females). Whereas HeLa cells gave rise to clinically detectable tumors in less than 20 days in 5/5 mice (one male, four female), no tumors formed in any of the mice injected with myoblast preparations (Fig. 4C–E). Accordingly, no human HLA-positive, laminpositive, or Ki-67-positive cells were detected in any of the mouse muscles analyzed at day 60 (Fig. 4F).
Discussion
This is the first report of the genomic safety profile of human myoblast preparations. Genomic integrity was investigated by several approaches including karyotype analysis, FISH, CGH array, and exome sequencing. Half of the myoblast preparations presented with normal karyotype, while the other half displayed a minor proportion of abnormalities, that is, 1–4 alterations/30 metaphases. It should be noted that, in preparations M3 and M7, mosaic aneuploidy was not confirmed by FISH, suggesting that the corresponding anomalies had occurred during the additional step of cell culture required to perform the karyotypic analysis rather than being present in the myoblast preparation itself. There was no recurrent karyotypic anomaly, except in 3/10 samples where chromosome 2 trisomy was found in 1–2/30 metaphases and/or 2/100 nuclei by FISH. The results of CGH array analyses suggested that no infra-karyotypic gene copy number variation had accumulated during culture above a frequency of approximately 30%, which is possibly due to the somewhat low sensitivity of CGH array to detect mosaic chromosomal changes. It was recently demonstrated that the consequences of aneuploidy on cells include induction of stress response genes and downregulation of cell cycle and cell proliferation genes (28). Our results further suggest that cells affected by chromosome alterations that unavoidably arise during cell cultures subsequently exit the cycle and presumably die. Specific investigations on the in vivo behavior of such cells would require obtaining clones or a cell line, which would be difficult given the lack of expansion observed in this study and the general difficulty of immortalizing myoblasts.
To our knowledge, none of the autosomal aneuploidies or structural changes observed at P3 has been described as constitutional karyotypic abnormalities in humans, reinforcing the view that they are lethal for the cell. Hence, a minority of myoblasts may exhibit transient, donordependent chromosomal alterations that do not confer any growth-selective advantage but are likely lethal for the cells in culture. The occurrence of myoblast chromosomal abnormalities evidenced in this study compares to pseudomosaicism, which for a long time has been known to occur in a minority of amniocytes when cultured to perform constitutional karyo typic analysis. In this context, trisomy 2 is also the most frequent artifact reported [for review see, (12)].
Our results are consistent with those of the recent report by Tarte et al. in which karyotypic abnormalities were observed in 25% of human MSC preparations (29). In the latter work, the frequency of karyotypic abnormalities, when present, was two to three aneuploidies/20–30 metaphases, except one outlier case presenting a mosaic trisomy 5 in 15/20 metaphases. Regarding MSCs, the report of spontaneous transformation in long-term cultures raised controversy in the field of cell therapy using adult stem cells (26). Unexpectedly, the reported phenomenon finally appeared to be secondary to a laboratory cross-contamination of cell cultures (9), which led to the retraction of the paper (6). Similarly, another group reported the possible occurrence of MSC transformation (25) before also concluding cross-contamination by other cell lines (30). Interestingly, none of the karyotypic abnormalities reported by Tarte et al. were associated with any growth advantage and MSCs with or without chromosomal alterations entered replicative senescence and did not exhibit any tumorigenic potential (29).
The risk of transformation of human myoblasts appears extremely low a priori, since this cell type is known to undergo progressive loss of its division capacities in prolonged cell culture, leading to cellular senescence (5). This is also what we observed here with our myoblast preparations. The lack of tumorigenic potential was further attested by a series of experimental evidence such as the behavior of myoblasts in soft agar culture, an assay in which only transformed cells can proliferate (33). There were no molecular anomalies in individual p53 transcripts, which otherwise constitute a prerequisite for cellular transformation. Consequently, no tumor arose after in vivo injection of myoblast preparations into immunodeficient mice, and as expected, when myoblasts are injected into normal noninjured muscle, there was even no engraftment of human cells in mouse muscle. The aim of the latter experiment was not to address the functionality of the myoblast preparation—which has been investigated elsewhere in a rat model of anal sphincter incontinence (1)—but to verify the lack of tumor development. It could be argued that scid mice may be leaky and thus partially immunocompetent, which could participate in the disappearance of the injected human cells. Yet, if myoblast preparations had contained overtly tumorigenic clones, they should have grown in vivo as the human HeLa control cells did in the same environment, even in the absence of muscle lesion, which is known to favor engraftment.
The origin of the genomic abnormalities observed herein remains elusive. It can be hypothesized that the cellular stress caused by in vitro manipulation and culture conditions contributes to their occurrence. This may imply the oxidative stress due to a higher oxygen pressure than in physiological conditions, the effect of growth factors, the use of trypsin, or even light exposure when manipulating the cells. Here we found no evidence for an obvious role of age or sex of the donor, but the sample size is too small to draw definitive conclusions. Owing to the rapid disappearance of cells bearing abnormalities, it is unlikely that they may have consequences on the fate of the cell product.
Together, the results of this study show that, inasmuch as MSCs, a minority of myoblasts may occasionally present nonrecurrent donor-dependent genomic abnormalities that randomly occur during the cell culture process but are not associated with a risk of transformation. Together with the fact that no tumor-related side effect has ever been reported in patients receiving injections of allogeneic (13,14,31,32) or autologous myoblasts (8,18,19,27), it is reasonable to conclude the safety of this cell therapy product. While we used deep molecular and cellular analyses for the necessity of this first description of the genomic stability of human myoblasts, we believe that, on a routine basis, it would not be justified to include sophisticated analyses such as CGH array, p53 functional assay, or exome sequencing in quality control. Furthermore, since karyotyping and FISH were not informative in our study and given that no threshold has yet been established to define quantitatively an abnormal level of aneuploidy for a cell therapy product, it is thus difficult to consider them currently as appropriate controls for the release of myoblast preparation for clinical use.
Previous clinical trials for Duchenne muscular dystrophy or chronic heart failure have used cell preparations containing on average 86% to 97% CD56+ cells (14,18,19,32). Heterogeneity of the cell product in terms of CD56+ content, for example, 67% to 97% for the study by Menasche et al. (19), may be reduced by the use of an additional step of anti-CD56 antibody-mediated purification, that is, 94% to 99.6% for the study of Karpati et al. (14). At this stage of the clinical development of myoblast transfer therapy, it is difficult to anticipate what could be an appropriate cutoff limit of CD56 positivity for myoblast preparations, and ongoing and future clinical trials will help solve this issue. In the case of urinary incontinence, Mitterberger et al. (22) used a mixture of myoblasts (CD56+) and fibroblasts (CD56-) based on the hypothesis that fibroblasts could exert beneficial effects. This is why, in the prospect of further developing myoblast therapy for fecal incontinence, we decided not to use a step of CD56+ enrichment. This yielded cell preparations with an average of 57% CD56+ cells in the present report. The heterogeneity of the cell product does not seem to be donor or age dependent. It is presumable that the CD56- cell population contains both myoblasts that have lost CD56 (when becoming locally confluent, for instance) and fibroblasts. The observed heterogeneity may originate, at least partly, from different initial proportions of CD56+ and CD56- cells and from different growth rates of these two cells types in vitro.
Based on the present product characterization and on proof-of-concept evidence that intrasphincteric injection of murine myoblasts is efficient in treating rats with fecal incontinence (1), a protocol for evaluating the clinical efficacy of autologous myoblasts produced with the same process in this report has been accepted by the French regulatory agency, and we have started a placebo-controlled clinical trial in patients with refractory fecal incontinence (ClinicalTrials.gov NCT01523522).
Footnotes
Acknowledgments
The authors would like to thank Jean-Jacques Tuech, Jean-Christophe Sabourin, Dominique Verschaeve, Arnaud Roucheux, Ingrid Dedreux, Geneviève Hacher, Reynald Publier, Angélique Aublet, Luc Sensébé, Vincent Mouly, Stéphane Le Mignot, Elise Fahler, Caroline Boyer, Marie Ponchon, Coralie Tiercin, Laurence Quirins, Marianne Colin, Marion Bougeard, Géraldine Maillet, Jérôme Couteau, and Sophie Coutant for their help during the study. This work was supported by Fondation de l'Avenir, Programme Hospitalier de Recherche Clinique and Association Nationale de la Recherche et de la Technologie. Aurélie Bisson benefited from a Ph.D. CIFRE fellowship funded in part by Celogos, and Christelle Doucet is an employee of Celogos. The authors declare no additional conflicts of interest. We are grateful to Nikki Sabourin-Gibbs, Rouen University Hospital, for editing the manuscript.