
Abstract
Serum is regarded as an essential supplement to promote survival and growth of cells during culture. However, the potential risk of transmitting diseases disqualifies the use of serum for clinical cell therapy in most countries. Hence, most clinical cell therapy programs have replaced human serum with human serum albumin, which can result in inferior quality of released cell products. Photochemical treatment of different blood products utilizing Intercept? technology has been shown to inactivate a broad variety of pathogens of RNA and DNA origin. The present study assesses the feasibility of using pathogen-inactivated, blood group-compatible serum for use in human pancreatic islet culture. Isolated human islets were cultured at 37°C for 3–4 days in CMRL 1066 supplemented with 10% of either pathogen-inactivated or nontreated human serum. Islet quality assessment included glucose-stimulated insulin release (perifusion), ADP/ATP ratio, cytokine expression, and posttransplant function in diabetic nude mice. No differences were found between islets cultured in pathogen-inactivated or control serum regarding stimulated insulin release, intracellular insulin content, and ADP/ATP ratio. Whether media was supplemented with treated or nontreated serum, islet expression of IL-6, IL-8, MCP-1, or tissue factor was not affected. The final diabetes-reversal rate of mice receiving islets cultured in pathogen-inactivated or nontreated serum was 78% and 87%, respectively (NS). As reported here, pathogen-inactivated human serum does not affect viability or functional integrity of cultured human islets. The implementation of this technology for RNA- and DNA-based pathogen inactivation should enable reintroduction of human serum for clinical cell therapy.
Introduction
From the very beginning of modern cell culture, serum has been regarded as an essential supplement to provide for survival and growth of cells isolated from different species and tissues (13). Nevertheless, the potential risk of transmitting different diseases disqualified the use of serum for clinical cell therapy in most countries. In spite of the increasing efficiency of pathogen screening protocols, the risk for transfusion-transmitted infections still persists and remains a major concern of public health care (55). Therefore, the majority of clinical cell therapy programs have replaced human serum with human serum albumin.
There are several pathogen inactivation techniques available to treat blood components; however, combined techniques are considered to provide the highest safety for patients (53). The Intercept? technology, routinely applied for pathogen inactivation of platelets and plasma, uses psoralen. Psoralen is comprised of small molecules able to pass through cell membranes and capsids, which subsequently bind to the helical regions of the nucleid acid. When UV-A light of 300–400 nm is emitted, psoralen is cross-linked to DNA and RNA either free or located in the genome, thus blocking both transcription and replication of viruses, bacteria, and protozoa. The efficiency and safety of this technology has been documented for clinical applications of different blood products (22,35,56). Preliminary studies demonstrate the principal suitability of this technology for clinical cell therapy by culturing human T lymphocytes, mesenchymal stem cells, and isolated pancreatic islets in media supplemented with pathogen-inactivated human serum (54).
The present study was initiated to evaluate the feasibility of pathogen inactivation for culturing isolated human islets of Langerhans. Although adult pancreatic islets are composed of different populations of highly differentiated cells that do not grow in vitro, the enormous metabolic demand of this tissue requires careful handling to maintain viability and integrity of these micro-organs in culture. In accordance with the majority of protocols for clinical islet transplantation, isolated human islets were cultured for a period of 3–4 days in CMRL 1066. The culture medium was supplemented with heat-inactivated human serum that was either untreated or pathogen inactivated by means of the Intercept® technology.
Materials and Methods
Pathogen Inactivation
Whole blood was collected from healthy donors and allowed to coagulate. Collected serum was pathogen inactivated utilizing the Intercept® technology (Cerus Europe BV, Amersfoort, Netherlands) as described for human plasma. Briefly, ABO-matched, freshly collected serum from six different healthy blood donors was mixed and split into two equal portions. While one aliquot was left untreated as control serum, the other portion was treated with the Intercept Processing Set for Plasma. The processing set utilizes a closed system consisting of a series of plastic containers. The serum aliquot (650 ml) was connected via a sterile connection devise and passed through a container of amotosalen hydrochloride (15 ml, 6 mM) into an illumination container and mixed thoroughly. The mixture was then exposed to 3 joule/cm2 UVA light (320–400 nm) from Intercept UVA Illuminator (Cerus). Afterwards, the UVA-treated serum was transferred through a compound adsorption device (CAD) to eliminate amotosalen residues and into a storage container. Both, control serum and pathogen-inactivated serum were then heat inactivated for 30 min at 56°C.
Islet Isolation
Local ethics committees approved all human studies. Research grade pancreata were procured from 12 brain-dead multiorgan donors within the Nordic Network for Clinical Islet Transplantation utilizing cold perfusion with either University of Wisconsin solution (ViaSpan©, DuPont Pharmaceuticals Ltd., Herts, UK) or Custadiol (Köhler Chemie GmbH, Alsbach, Germany). All pancreata were processed in a central isolation unit located in Uppsala, Sweden (49).
Islet isolation and purification was performed as previously described utilizing either Liberase HI (Roche, Indianapolis, IN, USA) or collagenase NB1 (Serva, Heidelberg, Germany) for pancreas digestion (16). Subsequent to purification and culture, islet yield and purity were determined in a standardized procedure utilizing a digital analysis system that discriminated dithizone-positive islets and nonstained particles (Cellimage, Uppsala, Sweden) (15).
Islet Culture
After purification, islets were precultured at 37°C in untreated single transfer packs for platelets (Baxter Medical AB, Stockholm, Sweden) (20) utilizing CMRL 1066 (Mediatech Inc., Manassas, VA, USA) supplemented with 10 mM HEPES (Invitrogen AB, Stockholm, Sweden), 10 mM nicotinamide (Swedish Pharmacy, Umeå, Sweden), 2 mM L-glutamine (Invitrogen), 50 μg/ml gentamicin (Invitrogen), 0.25 μg/ml fungizone (Invitrogen), 5 mM sodium pyruvate (Swedish Pharmacy), 20 μg/ml ciprofloxacin (Bayer, Leverkusen, Germany), and 10% blood group-compatible human serum. After 24–36 h medium was changed and aliquots of 1200 islet equivalents were transferred into untreated petri dishes (Sterilin, VWR, Stockholm, Sweden) and suspended into CMRL 1066 supplemented with 10% of either control human serum or pathogen-inactivated serum. Subsequent to 3–4 days of culture at 37°C islets were harvested and quality assessment was performed.
Islet Characterization
Stimulated insulin release of 20 equally distributed islets was measured in duplicate during dynamic glucose perifusion (Brandel, London, UK) in order to calculate the stimulation index, which was defined as the ratio between the areas under the curves that were calculated for the low (1.67 mM) and high (16.7 mM) glucose concentrations (20). In some preparations the low glucose values were close to zero, giving a very high stimulation index. Therefore, the maximum stimulation index for any islet preparation has been set to 30, as this is more physiologically realistic. The insulin content in the effluent, collected in 6-min intervals, was determined by using an enzyme immunoassay specific for human insulin (Mercodia, Uppsala, Sweden). The intracellular insulin content was measured in homogenized islets and normalized to the DNA content determined by means of the Quant-it Picogreen® assay (Invitrogen).
Homogenized islets were also assessed for expression of inflammatory mediators utilizing a Gyrolab?™ workstation (Gyros AB, Uppsala, Sweden). For each chemokine two monoclonal antibodies had to be loaded into a Gyrolab Bioaffy® 200 CD micro-laboratory disk to bind and detect tissue factor (TF, capture: TFE, Enzyme Research Laboratories, South Bend, USA; detection: CD 142, BD Pharmingen, Stockholm, Sweden), monocyte chemoattractant protein-1 (MCP-1, capture and detection: anti-CCL2/MCP-1 R&D Systems, Minneapolis, MN, USA), interleukin-6 (IL-6, capture: rat anti-human IL-6, BD Pharmingen; detection: anti-human IL-6, R&D Systems), or interleukin-8 (IL-8, capture and detection: anti-CXCL8/IL-8 R&D Systems). An aliquot of 80 islets was used to measure the ADP/ATP ratio utilizing the ApoGlow kit (Cambrex Bio Science, Nottingham, UK).
Islet in vivo function was assessed in NMRI nude mice (Scanbur AB, Sollentuna, Sweden) rendered diabetic by a single intravenous injection of 90 μg/g alloxan (Sigma-Aldrich AB, Stockholm, Sweden) 3 days prior to transplantation of an aliquot of 3000 islet equivalents beneath the kidney capsule. Blood samples were taken from the recipients' tail vein every 2–4 days and analyzed utilizing a one-touch glucometer (Bayer, Leverkusen, Germany). Pretransplant, the nonfasting serum glucose levels of all recipients exceeded 400 mg/dl. After transplantation nonfasting serum glucose levels <200 mg/dl were defined as normoglycemic and considered as graft function. Thirty-five days posttransplant nephrectomy of graft-bearing kidneys was performed to confirm the functional dependency of normoglycemia on the transplanted islets.
Statistical Analysis
Statistical analysis was performed utilizing Prism 5.0c (GraphPad, La Jolla, CA, USA). Comparison between experimental groups was carried out by the Wilcoxon test for matched samples and Fisher's exact test. Data are presented as medians with range except for dynamic insulin release where data are presented as means ± SEM. Differences are considered significant at p < 0.05. Values of p > 0.05 are termed nonsignificant (NS).
Results
Islet Characterization
As shown in Figure 1, the amount of insulin secreted during glucose perifusion was similar in both experimental groups. Also, the incubation in culture medium supplemented with either pathogen-inactivated or control serum did not influence the dynamics of insulin release. The median stimulation index differed only marginally between islets cultured in either pathogen-inactivated 11.05 (range 1.2–30) or untreated serum 19.1 (range 1.1–30, NS, p = 0.57).
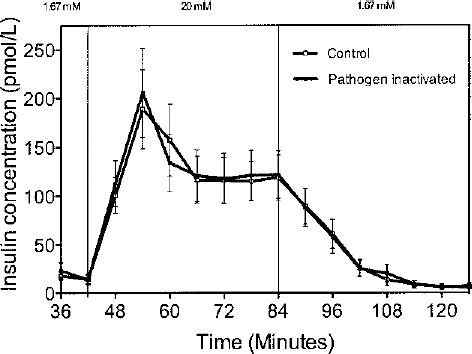
Dynamic insulin release measured during glucose perifusion of isolated human islets cultured for 3–4 days at 37°C in CMRL 1066 supplemented with 10% of either untreated control serum (open squares) or pathogen-inactivated (filled squares) serum. Values are expressed as mean ± SEM from 12 different islet preparations.
Additionally, no difference was observed regarding the median intracellular insulin content of islets cultured for 3–4 days in pathogen-inactivated 1.45 (range 0.19–10.55 ng/ng DNA) or control serum 1.45 (range 0.22–7.96 ng/ng DNA, NS) (Fig. 2A).
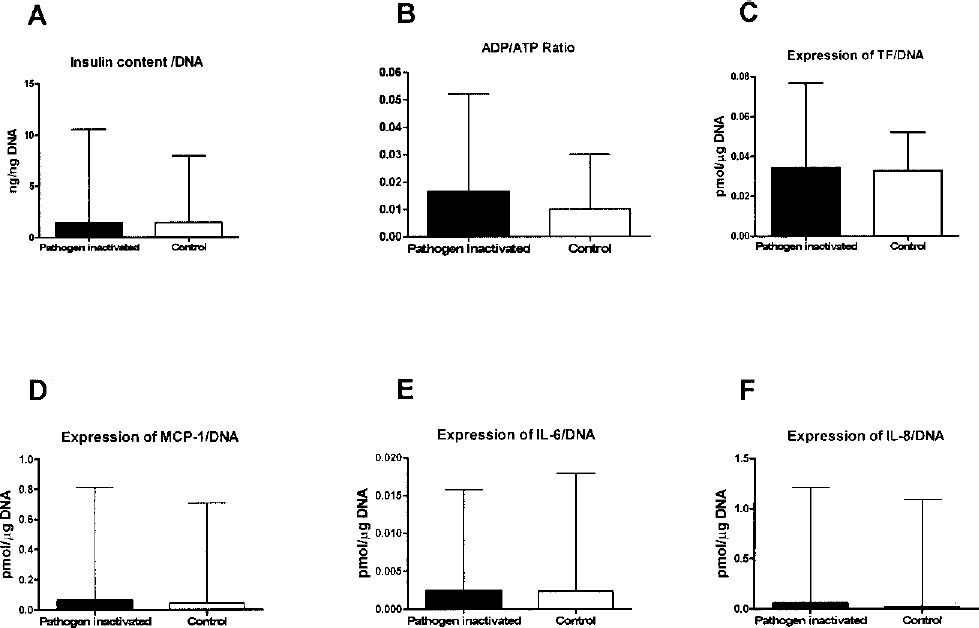
Human islet characterization after 3–4 days of 37°C culture in CMRL 1066 supplemented with 10% of either untreated control serum (open bars) or pathogen-inactivated serum (filled bars). Characterization included (A) intracellular insulin content, (B) ADP/ATP ratio, and expression of (C) tissue factor (TF), (D) MCP-1, (E) IL-6, and (F) IL-8. Data are expressed as median with range from 12 different islet donors.
As depicted in Figure 2B, the median ADP/ATP ratio was slightly higher in islets cultured in pathogen-inactivated serum 0.016 (range 0.01–0.052) when compared to standard culture conditions 0.010 (range 0.01–0.30). However, this difference did not reach statistical significance (p = 0.26).
Supplementation with pathogen-inactivated serum did not influence the expression of different chemokines in cultured islets as displayed in Figure 2 C-F. The median expression levels of tissue factor [0.034, range (0.015–0.076) vs. 0.033 (range 0.011–0.052) fmol/?g DNA, NS] (Fig 2C), MCP-1 [0.064 range (0.011–0.82) vs. 0.045 (range 0.010–0.71) fmol/μg DNA, NS] (Fig. 2D), IL-6 [0.0024 (range 0.0007–0.016) vs. 0.0024 (range 0.0006–0.018) fmol/μg DNA, NS] (Fig. 2E), and IL-8 [0.058 (range 0.0003–1.210) vs. 0.0023 (range 0.0002–1.090) fmol/μg DNA, NS] (Fig. 2F) were not affected by the use of pathogen-inactivated serum to that of control serum.
Diabetic Nude Mice Bioassay
Potency of cultured islets was assessed after transplantation beneath the kidney capsule of diabetic nude mice. In concordance with the findings made for islet in vitro function, reduction of hyperglycemia was similar in islet recipients of either experimental group (Fig. 3). The final cure rate of mice receiving islets cultured in pathogen-inactivated or nontreated serum was 7 (78%) and 8 out of 9 (87)%, respectively (NS by Fisher's exact test).

Graft function of cultured human islets transplanted beneath the kidney capsule of alloxan-treated NMRI nude after 3–4 days of 37°C culture in CMRL 1066 supplemented with 10% of either (A) untreated control serum (n = 9, open symbols) or (B) pathogen-inactivated serum (n = 9, filled symbols). Graft removal through nephrectomy (Nx) was performed as indicated at day 35 posttransplant. Presented data are from three different islet donors.
Discussion
Pathogen inactivation with the Intercept? technology was assessed on human serum used for culture of human islets of Langerhans in a clinical setting.
Human islets isolated for clinical purposes are cultured primarily for logistical reasons. Most clinical islet transplant protocols include a culture period of 2–4 days to facilitate pretransplant tissue quality assessment and islet anti-inflammatory pretreatment, recipient matching for transplantation, and induction of immunosuppressive protocols (6,24,39). In addition, different experimental studies indicated that in vitro culture reduces islet immunogenicity compared to freshly isolated islets (27,29,32). A period of culture has benefits not only for islet recovery, as it replenishes islet ATP stores depleted during both cold ischemia period and the isolation procedure, but also increases posttransplant function of human islets transplanted into diabetic nude mice (2,5,23,26).
Supplementation of culture medium with serum seems to be important to preserve the functional and morphological integrity of islets. Serum, if added to a suitable culture medium, prevents fragmentation of cultured islets and preserves the secretory capacity of β-cells of different species during culture (1,25). This effect seems to be particularly pronounced if serum of the donor species is used for supplementation in comparison to xenogenic serum (4,18).
Although it was previously demonstrated that a minimum percentage of serum is required to preserve functional islet integrity (1,7), a few studies reported that serum-free culture media are superior to serum-supplemented media for function of cultured human islets (3,14). This finding can be related to the observation that human islets are less sensitive toward serum deprivation than islets of other species (40). Nevertheless, serum-free media have to be supplemented with numerous growth factors and nutrients to preserve survival and function of cultured human islets (17,48). Among these additives, serum albumin is considered to be a component that can partially replace serum (33,38). More recent reports clearly emphasize the importance of supplementation of culture media with complete serum compared to human serum albumin because it improves human islet survival, function, and viability (30,43). These observations are in agreement with basic approaches clearly suggesting that the complexity of serum components does not justify the assignment of different growth-promoting abilities to individual protein fractions such as albumin (19).
Nevertheless, in spite of the obvious advantages to use serum for islet cell culture this essential supplement is in most centers avoided in clinical cell therapy to reduce the risk of transmitting diseases by blood components. The threat to public health concerns particularly the spread of HIV, HCV, HBV, and HTLV. Also, the transmission of bacteria and protozoa still represents significant risks associated with therapeutic utilization of blood components (31).
Although extensive blood donor testing has dramatically reduced the risk of contamination of blood components, transfusion-transmitted infections are still reported. Moreover, the appearance of new pathogens in the population remains a major concern as seen with West Nile virus in North America and the SARS virus in Asia (12,44).
The Intercept® technology using amotosalen and UVA treatment, already in use in many European countries (50), has been reported to be safe in preclinical studies (9–11) and to be well tolerated in a wide range of patients (22,41,42,45,52,56). It has even been used to prevent transfusion transmission of Chikungunya virus during an epidemic (46). The Intercept® technology has proven its efficacy towards bacteria, viruses, and protozoan parasites (8,21,28,34–37,46,47,51). Implementation of this safe and validated method for pathogen inactivation using Intercept® can provide protection from both known and yet unidentified RNA- and DNA-dependent agents.
In conclusion, the present data suggest that human serum treated by Intercept® technology can be used for human islet cell culture without affecting viability and functional integrity. The implementation of this technique for prevention of pathogen transmission would allow for the broad reintroduction of the use of human serum for clinical cell therapy worldwide.
Footnotes
Acknowledgments
This study was supported by grants from Juvenile Diabetes Foundation International, the National Institutes of Health, Vinnova Sampost, and the Swedish Research Council. The authors would like to thank Anna Karlsson and Elisabeth Wijkström for their excellent work with the Gyros system, Karin Andersson for her skilled work transplanting mice, and Karin Leijskog for providing both control and pathogen-inactivated serum. M.U.S. performed the experiments and participated in islet isolation, data analysis, and writing the paper. D.B. participated in islet isolation, data analysis, and writing the paper. O.K. participated in research design and writing the paper. F.K. participated in research design and writing the paper.