
Abstract
The success of liver cell therapy remains closely dependent on how well the infused cells can be accepted after transplantation and is directly related to their degree of immunogenicity. In this study, we investigated the in vitro immunogenic properties of isolated human hepatocytes (hHeps) and adult-derived human liver progenitor cells (ADHLPCs), an alternative cell candidate for liver cell transplantation (LCT). The constitutive expression of immune markers was first analyzed on these liver-derived cells by flow cytometry. Human liver-derived cells were then cocultured with allogeneic human adult peripheral blood mononuclear cells (PBMCs), and the resulting activation and proliferation of PBMCs was evaluated, as well as the cytokine levels in the coculture supernatant. The effect of liver-derived cells on monocyte-derived dendritic cell (MoDC) properties was further analyzed in a secondary coculture with naive CD4+ T-cells. We report that hHeps and ADHLPCs expressed human leukocyte antigen (HLA) class I and Fas but did not express HLA-DR, Fas ligand, and costimulatory molecules. hHeps and ADHLPCs did not induce T-cell activation or proliferation. Moreover, hHeps induced a cell contact-dependent production of interleukin (IL)-10 that was not observed with ADHLPCs. The IL-10 was produced by a myeloid DC subset characterized by an incomplete mature state. Furthermore, hHep-primed MoDCs induced an antigen-independent hyporesponsiveness of naive CD4+ T lymphocytes that was partially reversed by blocking IL-10, whereas nonprimed MoDCs (i.e., those cultured alone) did not. hHeps and ADHLPCs present a low immunogenic phenotype in vitro. Allogeneic hHeps, but not ADHLPCs, promote a cell contact-dependent production of IL-10 by myeloid DCs, which induces naive CD4+ T-cells antigen-independent hyporesponsiveness.
Introduction
Liver cell transplantation (LCT) has already shown promising results in restoring metabolic function in patients (17,41,42,44,45). However, the success of the technique remains closely related to the fate of the liver cells after transplantation.
The liver is widely considered as an immunoprivi-leged organ that can favor the induction of immunologic unresponsiveness or even tolerance (36). Liver tolerance has been highlighted by several lines of evidence, such as the lack of systemic inflammation despite a continuous exposure to foreign antigens from the portal circulation (12), the relatively low occurrence of T-cell-mediated rejection in liver transplant recipients and, in some cases, the acceptance of liver grafts despite the absence of an immunosuppressive regimen (30), as well as the demonstration of the liver transplant's ability to facilitate the acceptance of other grafted organs (11).
Unexpectedly, mouse liver cells appear to be highly immunogenic and can be rejected by different immune mechanisms than other transplanted organs (20). Indeed, several in vivo studies in murine hepatocyte transplantation models demonstrated the role of T-cells in the hepatocyte allograft rejection implicating both cluster of differentiation 4-positive (CD4+) and CD8+ T-cell-dependent pathways (7). The contribution of costimulatory signals has also been shown to be determinant in this alloimmunity (18). So far, few studies have been performed with isolated human liver cells in the context of clinical cell transplantation, and more data are needed to determine the immunogenicity of these human cells.
T-cell activation in response to alloantigens is mediated through T-cell receptor (TCR) interaction with pep-tides presented in the context of human leukocyte antigen (HLA) molecules on antigen-presenting cells (APCs). Within the APC family, dendritic cells (DCs) represent a professional subset present in virtually all organs. DCs can induce immunostimulatory as well as immunoregula-tory responses depending on their ontogeny, state of differentiation, or their maturation (4). In transplantation, DCs play a key role in the regulation and, especially, in the polarization of alloreactive T lymphocytes (47). In the field of murine hepatocyte transplantation, Bumgardner et al. have shown the involvement of APC-related costimulatory signals in cell rejection, underlining the importance of APCs in this process (9).
In the present study, we investigated the in vitro immunogenic properties of human hepatocytes (hHeps) from postmortem donor livers and compared them to those of adult-derived human liver progenitor cells (ADHLPCs). ADHLPCs represent an in vitro expandable cell source currently being developed for regenerative liver medicine, with a high proliferative capacity, an ability to differentiate in vitro and in vivo into functional hepatocyte-like cells, and a mesenchymal phenotype that could potentially be associated with a different immunologic profile compared to hHeps (32). In this context, we first analyzed the constitutive expression of cell surface immunologic markers. We then evaluated the capacity of hHeps and that of ADHLPCs to induce an immune response after coculture with allogeneic human adult peripheral blood mononuclear cells (PBMCs). Finally, we focused on the properties of monocyte-derived DCs (MoDCs) induced by hHeps, in particular, the ability of MoDCs to modulate the immune response.
Materials and Methods
Cell Isolation and Culture
The present study was approved by the local ethics committee. Written informed consent was obtained from each individual before blood sampling for PBMC isolation or from next of kin for cadaveric donor livers.
Human Hepatocyte Isolation Procedure and Culture
hHeps (n = 22) were isolated from healthy livers of cadaveric donors (see Table 1A) using the two-step collagenase perfusion technique. Liver isolation and hHeps cryopreservation/thawing procedures were previously published in detail (32,40). Cell viability was estimated by trypan blue (Invitrogen, Merelbeke, Belgium) exclusion.
General Characteristics of Liver-Derived Cell Donors

Abbreviations: ADHLPC, adult-derived human liver progenitor cells; ND, not determined.
For cell culture, hHeps were plated at 2 × 105 cells/cm2 on collagen I-coated plates (BD Biosciences, Erembodegem, Belgium) in Williams' E medium (Invitrogen, Merelbeke, Belgium) supplemented with 10% fetal calf serum (FCS; A&E Scientific, Marcq, Belgium), 25 ng/ml epidermal growth factor (EGF; Peprotech, London, UK), 10 mg/ml insulin (Lilly, Brussels, Belgium), and 1% of penicillin/streptomycin (Invitrogen). Where specified, interferon (IFN)-γ (100, 500, 1,000 UI/ml; R&D Systems, Abingdon, UK) or tumor necrosis factor (TNF)-α (100, 500, 1,000 UI/ml; R&D Systems) was added to the culture. After 24 to 96 h of culture, cells were harvested using trypsin with 0.05% ethylenediaminetetraacetic acid (EDTA; Invitrogen), washed in Dulbecco's phosphate-buffered saline (DPBS; Westburg, Leudsen, The Netherlands) to eliminate cell debris, and resuspended in DPBS. The viable cell number of the single cell suspension thereby obtained was determined by the trypan blue dye exclusion method. One hundred thousand live cells were then stained and analyzed by flow cytometry (see later section). The analysis was performed after gating on the live cell population on the forward scatter (FSC)/side scatter (SSC) dotplot.
Progenitor Cell Preparation and Culture
ADHLPCs (n = 9) were isolated from healthy livers of cadaveric donors (see Table 1A), except for one specimen isolated from a living donor with a metabolic defect, and cultured as previously described (32). Cells were used from third to sixth passages. Whenever possible, ADHLPCs were compared to hHeps obtained from the same donor. ADHLPCs were plated at 1 × 104 cells/cm2 on collagen I-coated plates (BD Biosciences) in Dulbecco's modified Eagle's medium (DMEM) with high glucose concentration (Invitrogen) supplemented with 10% FCS and 1% of penicillin/streptomycin. Cells were processed following the same procedure as for hHeps.
PBMC Isolation
PBMCs (n = 17) were isolated from the buffy coat leukocytes of healthy adult anonymous volunteers (Belgian Red Cross, Namur Blood Center, Namur, Belgium) by density gradient centrifugation using Ficoll-Paque (GE Healthcare, Diegem, Belgium). Cell viability estimated by trypan blue exclusion was >95%.
PBMC Carboxyfluorescein Diacetate Succinimidyl Ester (CFSE) Staining
Carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) was dissolved to 5 mM with 18 μl of dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA) according to the manufacturer's instructions. Sixty microliters of 5 mM stock CFSE solution was added to 8 × 106 PBMCs suspended in prewarmed PBS/bovine serum albumin (BSA; Sigma) 0.1% at a final concentration of 8 × 106 cells/ml. Cells were incubated at 37°C for 10 min. The staining was quenched by the addition of five volumes of ice-cold culture media to the cell suspension. PBMCs were incubated for 5 min on ice and then centrifuged at 400 × g for 20 min at 4°C. Cells were resuspended in fresh medium and washed three times.
Generation of Human Monocyte-Derived Dendritic Cells (MoDCs)
The CD14+ fraction of PBMCs was collected by immunomagnetic-positive selection using the CD14+ cell isolation kit with magnetic-activated cell sorting (MACS) columns (MiltenyiBiotec, AD Utrecht, The Netherlands) and subsequently cultured in Roswell Park Memorial Institute (RPMI)-1640 (Gibco BRL, Merelbeke, Belgium) medium supplemented with 1% of penicillin/streptomycin (Invitrogen), 2 mM l-glutamine (GibcoBRL), 1% nonessential amino acids (Gibco BRL), 10% heat-inactivated FCS (Hyclone, Erembodegem, Belgium), 50 μM β-mercaptoethanol (Gibco BRL), 800 U/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF; Peprotech), and 200 U/ml of interleukin (IL)-4 (R&D Systems). Every 2 days, 800 U/ml of GM-CSF and 200 U/ml of IL-4 were added. After 6 days of culture, MoDCs were harvested, washed, and used for subsequent experiments. The resulting cell preparation contained more than 85% MoDCs as assessed by morphology and fluorescence-activated cell sorting (FACS) analysis (Lin-HLA-DR+ CD11c+; see later section).
Naive CD4+ T-Cell Isolation
The naive CD4+ T-cell population of PBMCs was collected by immunomagnetic negative selection using the naive CD4+ T-cell isolation kit with MACS columns (MiltenyiBiotec). Naive CD4+ T-cells were more than 90% pure (for CD4+, CD45RA+) as assessed by FACS analysis (see later section).
Mixed Lymphocyte Reaction (MLR)
Isolated PBMCs (105 cells) were used as responding cells and were cocultured with γ-irradiated allogeneic stimulating PBMCs (105 cells) in a 96-well plate (Greiner, Wemmel, Belgium) in RPMI-1640 medium for 7 days. The cell viability of γ-irradiated PBMCs estimated by blue trypan exclusion was >90%. Cells were pulsed with [3H]thymidine (1 μCi/well; MP Biomedicals, Irvine, CA, USA) 18 to 24 h prior to the end of the incubation, and the radioactive uptake was then measured with a TopCount NXT microplate scintillation and luminescence counter (Perkin Elmer, Zaventem, Belgium).
Liver-Derived Cells and PBMC Coculture Preparation
hHeps or ADHLPCs were seeded on collagen I-coated 24-well plates (BD Biosciences), allowed to adhere overnight, and cocultured at varying ratios with allogeneic PBMCs separated or not by a semipermeable membrane (ThinCert, 0.4 μm; Greiner Bio-One, Wemmel, Belgium) in RPMI-1640 medium. After 24 to 96 h of coculture, the supernatants were collected and stored at −20°C for cytokine level detection by enzyme-linked immunosor-bent assay (ELISA; see later section). In order to analyze the PBMC activation and the intracellular cytokine production, brefeldin A (10 mg/ml; Sigma) was added to the coculture for the last 4 h of incubation. After 24, 48, 72, and 96 h of coculture, PBMCs were harvested, stained, and then analyzed using flow cytometry. The proliferative response of PBMCs (105 cells/well in a 96-well plate) to liver cells was evaluated by the [3H]thymidine incorporation method and by CFSE staining after 7 days of coculture with γ-irradiated hHeps or ADHLPCs (5 × 104, 2 × 104, or 104 cells/well). An allogeneic MLR was used as a positive control. The culture medium was changed every other day over the 7-day period to avoid depletion of arginine and growth factors. Cells were pulsed with [3H]thymidine (1 μCi/well), and the radioactive uptake was measured 18 to 24 h later with a TopCount NXT microplate scintillation and luminescence counter. The induced proliferation of both CD4+ and CD8+ populations was analyzed after CFSE staining by flow cytometry (see later section).
CD4+ T-Cell Stimulation
MoDCs were cultured alone or cocultured with allogeneic hHeps or ADHLPCs at the ratio 2:1 on collagen I-coated 24-well plates in RPMI-1640 medium. After 48 h, cells were harvested and MoDCs were isolated by immunomagnetic-positive selection using the dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) isolation kit with MACS columns (MiltenyiBiotec). MoDCs, hHep-primed MoDCs, or ADHLPC-primed MoDCs were then cocultured in 96-well plates with allogeneic naive CD4+ T-cells (2 × 105 cells) at different ratios in RPMI-1640 medium. The naive CD4+ T-cell proliferation was evaluated on day 7 after 18 to 24 h of [3H]thymidine addition. After 7 days, the naive CD4+ T-cells were harvested and stained for intracellular forkhead box P3 (Foxp3) expression. Where specified, a blocking antibody directed to IL-10 (10 μg/ml; R&D Systems) was added to the naive CD4+ T-cell coculture on day 0 and day 3. Mouse immunoglobulin G2B (IgG2B; R&D Systems) was used at the same concentration as an isotype control.
CD4+ T Restimulation Assay
CD4+ T-cells were collected after 7 days of primary coculture with MoDCs or hHep-primed MoDCs, recounted, and then restimulated by MoDCs from the same donor as the primary coculture or from an unrelated donor in a 96-well plate in RPMI-1640 medium. After 7 days of restimulation, CD4+ T-cell proliferation was evaluated by the [3H]thymidine incorporation method.
Flow Cytometry Analysis
The following antibodies were used for cell surface marker analysis: phycoerythrin (PE)-anti-HLA-ABC IgG2, allophycocyanine (APC)-anti-CD95 (Fas; Fas cell surface death receptor) IgG1 (all from BioLegend, Antwerp, Belgium), peridinin-chlorophyll protein (PerCP)-anti-HLA-DR IgG2, APC-anti-CD40 IgG1, PE-anti-CD80 IgG2, PE-cyanine 7 (Cy7)-anti-CD45 IgG1, PE-anti-CD73 IgG, APC-anti-CD90 IgG, fluorescein isothiocyanate (FITC)-anti-CD105 IgG2, APC-Cy7-anti-CD3 IgG1, PerCP5.5-anti-CD4 IgG1, PE-Cy7-anti-CD8 IgG1, FITC-anti-CD16 IgG1, FITC-anti-CD56 IgG2, PE-anti-CD16 IgG1, PE-anti-CD56 IgG1 PE-anti-CD19 IgG1, APC-anti-CD45 IgG1, PE-Cy7-anti-CD11c IgG1, FITC-anti-CD14 IgG2, PE-Cy7-anti-CD25 IgG1, PE-anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4; CD152) IgG2, PE-Cy7-anti-CD69 IgG1, FITC-anti-lineage 1 (Lin1) IgG cocktail (all from Becton-Dickinson, Erembodegem, Belgium), and FITC-anti-CD45RA+IgG2B (Miltenyi). The following antibodies were used for intracytoplasmic marker analysis: APC-anti-IL-10 IgG2, PE-anti-CD40 ligand IgG1, FITC-anti-IFN-γ IgG1, FITC-anti-CD40 ligand IgG1, PE-anti-IL-10 IgG2 (all from Becton-Dickinson), APC-anti-human Foxp3 (ImmunoSource, Schilde, Belgium). Cells were washed in PBS supplemented with 1% BSA and incubated for 20 min at 4°C with the antibodies or their corresponding isotype in 10% pooled heat-inactivated human serum. PBMCs were used as a positive control for HLA I staining, MoDCs were used as a positive control for HLA II staining, PBMCs treated with phytohemagglutinin (PHA; 5μg/ml for 48 h; Sigma) were used as a positive control for Fas (Fas cell surface death receptor; CD95) staining, and a lymphoblastic cell line [LCL; generated by infecting PBMCs from a male liver graft patient with Epstein-Barr virus (EBV)] was used as a positive control for CD40 and CD80 staining. Intracytoplasmic staining was performed by incubating the cells in 200 μl Cytofix/Cytoperm (BD Bioscience) for 20 min at room temperature (RT). Cells were washed with Perm/Wash solution (BD Bioscience) and centrifuged for 7 min at 400 × g. Cell pellets were resuspended in 50 μl Perm/Wash containing the antibodies or their corresponding isotype and incubated for 20 min at RT. For Foxp3 staining, cells were incubated with 1 ml of Fix/Perm solution (Immunosource, Schilde, Belgium) for 45 min at 4°C in the dark. The cells were then washed twice with 2 ml of permeabilization solution (Immunosource) and incubated with 20 μl of APC-anti-Foxp3 antibody (Immunosource) or the corresponding rat IgG2a APC isotype control antibody (BD Bisociences) for 30 min at 4°C in the dark. Cells were then washed, resuspended in CellFix (BD Bioscience), and analyzed on a FACS Canto II Flow Cytometer (Becton-Dickinson) using the FACS Diva software (Becton-Dickinson).
Hepatogenic Differentiation
ADHLPCs (n = 3) at passages 3, 4, and 6 were seeded at a density of 1 × 104 cells/cm2 in collagen I-coated 175-cm2 flasks (Greiner) and cultured using DMEM supplemented with 10% FCS (A&E Scientific) and 1% of penicillin/streptomycin (Invitrogen). Twenty-four hours later, the culture medium was switched to Iscove's modified Dulbecco's medium (IMDM; Invitrogen). Cells were incubated for 2 days with IMDM containing 20 ng/ml epidermal growth factor (EGF; Peprotech) and 10 ng/ml basic fibroblast growth factor (bFGF; Peprotech). Cells were then subjected to a three-step differentiation protocol. First, the cells were incubated for 10 days with IMDM containing 20 ng/ml hepatocyte growth factor (HGF; Peprotech), 10 ng/ml bFGF, nicotinamide 0.61 g/L (Sigma), and 1% insulin—transferrin—selenium (ITS; Invitrogen) premix. Then, the cells were incubated for 10 days with IMDM containing 20 ng/ml HGF, 20 ng/ml oncostatin M (Peprotech), nicotinamide 0.61 g/L, and 1% ITS premix. Finally, the cells were treated with IMDM containing 20 ng/ml oncostatin M, 1 μM dexamethasone (Sigma), and 1% ITS premix for 10 days. For each step, the medium was changed every 3 days. Negative controls were performed with cells cultured in IMDM supplemented with 1% FCS and 1% of penicillin/streptomycin.
Immunohistochemistry
Fresh human liver samples (n = 3; two males, one female; 16, 18, and 37 years old) were immediately snap frozen, embedded in optimum cutting temperature (OCT) compound (Sakura Finetek, Villeneuve d'Ascq, France), and stored at −80°C. Frozen liver sections (5 μm) were cut with a cryostat (Leica, Diegem, Belgium), fixed with acetone (VWR, Leuven, Belgium) for 10 min at RT, and then washed in PBS. The endogenous peroxidase activity was inhibited by a bath of methanol/hydrogen peroxide 0.3% (Sigma) for 15 min. Sections were then washed with distilled water. Nonspecific binding was blocked by incubating sections for 30 min at RT with PBS containing 5% of BSA. Tissues were incubated overnight with 1/200 mouse monoclonal anti-human HLA-ABC (DakoCytomation, Heverlee, Belgium), 1/200 mouse monoclonal anti-human HLA-DP, DQ, DR (DakoCytomation), and 1/200 mouse monoclonal anti-human CD95 (Biolegend). Sections were rinsed three times with PBS containing 0.1% Tween 20 (Acros, Illkirch, France) and then incubated 30 min with anti-mouse Dako Envision horseradish peroxidase (HRP) antibody (DakoCytomation). Staining was performed by 3-min exposure to diaminobenzidine (Sigma). After rinsing, nuclei were colored with Mayer's hematoxylin (Sigma), and cells were mounted. Negative experimental controls were performed (absence of primary antibodies). Sinusoidal endothelial cells from the fresh human liver samples were used as a positive control for HLA class II staining.
Immunocytochemistry
Cells isolated from human livers were cultured on a collagen I-coated glass coverslip (Fischer Scientifique, Tournai, Belgium) and fixed after 24 h with 3.5% formaldehyde (Sigma) for 15 min at RT. Cells were incubated with 3% hydrogen peroxide (Sigma) for 3 min and then washed with distilled water. Cells were permeabilized with 1% Triton X-100 (Sigma). After 1-h blockade in 1% BSA, cells were incubated for 1 h at RT in 0.1% BSA with primary mouse antihuman monoclonal antibodies as follows: 1/50 vimentin, 1/50 albumin (both Sigma), 1/50 α-smooth muscle actin (ASMA), 1/50 hepatocyte paraffin 1 (HepPar1; both DakoCytomation), 1/50 cytokeratin 18 (CK18; Progen, Heidelberg, Germany), 1/50 CK8 (Chemicon, Temecula, CA, USA), 1/20 dipeptidylpeptidase-IV (DPPIV; Ancell, Bayport, MN, USA), and with rabbit anti-human α-fetoprotein (αFP; DakoCytomation) polyclonal antibodies at 1/100. Cells were washed with PBS and exposed for 30 min with anti-mouse or anti-rabbit Dako Envision HRP antibody. Staining was performed by 3-min exposition to diaminobenzidine. After rinsing, nuclei were colored with Mayer's hematoxylin, and cells were mounted for analysis. Negative experimental controls were performed by omitting primary antibodies. The estimation of positive cells for the corresponding antibodies was carried out in three fields under 100× magnification for each specimen and was expressed as the average percentage of positive cells out of the total number of cells.
Cytokine Quantification
IL-6, IL-10, TNF-α, and IFN-γ were measured in the hHep suspension and the cell coculture supernatants by ELISA using antibodies from Biosource Europe (detection limit at 15 pg/ml; Invitrogen), according to the manufacturer's instructions, and analyzed with a Multiskan EX absorbance reader (Thermo, Erembodegem, Belgium).
Statistical Analysis
Results are expressed as mean±standard deviation (SD), as mean±standard error of the mean (SEM), or as median and range. Data were compared using the one-way ANOVA analysis with Bonferroni post hoc test. Results were considered as statistically significant with p < 0.05.
Results
The general characteristics of liver-derived cell donors are shown in Table 1A. HLA typing is given in Table 1B. Cell viability estimated by trypan blue exclusion was >70%, which is in accordance with the viability of cells previously used in LCT (45).
HLA Typing of the Liver-Derived Cell Donors

HLA, human leukocyte antigen; A, B, C class I; DP, DQ, DR class II.
Composition of the hHep Suspensions
We first investigated the composition of the cell suspension obtained after digestion of a liver under clinical protocols. The cell population subsets, the expression pattern of surface markers, and the cytokine profile were analyzed at the basal level. Freshly isolated hHep suspensions were mainly constituted by hepatocytes as demonstrated by immunocytochemistry (Fig. 1). A majority of cells (>95%) highly expressed hepatocyte-specific markers, such as albumin, αFP, CK 8 and 18, HepPar1, and DPPIV, while a very small proportion of cells (<1%) showed a positive staining for mesodermal antigens (vimentin and ASMA) demonstrating the presence of nonparenchymal cells in the suspension. Using flow cytometry, a subset population in the CD45-negative fraction that was positive for CD105, CD73, and CD90 was detected in all specimens with a median (range) of 0.1% (0.0-1.9) attesting the presence of mesenchymal cells (Table 2A). These results corroborated with the presence of positive cells for mesodermal antigens in the suspension since mesenchymal cells were also shown to express ASMA and vimentin. Moreover, a small subset of CD45-positive cells (values under 5% of positive cells were considered not significant) was detected in four of 14 freshly isolated hHep suspensions at 7.3% (6.4-26.3) [median (range)]. Complementary experiments demonstrated the presence of natural killer (NK) cells (CD16+ and CD56+) and APCs (CD11c+ or CD14+, CD80+) in the CD45+ subset. These contaminating cells were also observed after the cryopreservation/thawing procedure (Table 2B). Finally, we were not able to detect any significant level of cytokines (IL-6, IL-10, IL1-β, TNF-α, IFN-γ) in the freshly isolated or cryopreserved/thawed hHep suspensions using ELISA (data not shown).
Contaminating Cells in Human Hepatocyte Suspensions

Cell Subsets in the CD45 Positive Population Contaminating Human Hepatocyte Suspensions

Contaminating cells in human hepatocyte (hHep) suspensions were evaluated by flow cytometry after staining with the corresponding monoclonal antibodies. Data are expressed as percent of positive cells and presented as median (range) of 12 to 14 independent experiments. The presence of cluster of differentiation 45 positive (CD45+) cells >5% in the hHep suspension was considered as a significant contaminating population. Abbreviations: freshly hHep, freshly isolated human hepatocytes; cryohHep, cryopreserved/thawed human hepatocytes.

Analysis of the expression of hepatic and mesenchymal markers by human liver cells using immunohistochemistry. The composition of the cell suspension obtained after digestion of a liver under clinical protocols was investigated by immunocytochemistry. Expression of surface markers specific for human hepatocyte: (A) albumin, (B) α-feto-protein, (C) cytokeratin 8, (D) cytokeratin 18, (E) hepatocyte paraffin 1 (HepPar1), and (F) dipeptidylpeptidase IV. Expression of mesenchymal antigens: (G) vimentin and (H) α-smooth muscle actin. Insets show enlargement of vimentin (G) and α-smooth muscle actin (H) staining. Control staining performed without primary antibody is represented in (I). Picture magnification is 100×. One representative experiment out of eight is shown. Scale bar: 50 μm.
Human Hepatocytes and Liver Progenitor Cells Express MHC Class I and CD95 But Not HLA-DR or Costimulatory Molecules
As hHeps were the predominant cell type in the hHep suspensions, we further characterized their immunophenotype by analyzing the expression of specific cell surface markers at the basal level and after 24 to 96 h of incubation with either TNF-α or IFN-γ at graded concentrations (100-1,000 IU/ml) using flow cytometry. In freshly isolated hHep suspensions, viable hHeps were selected according to FSC and SSC parameters and were negatively stained for CD45. Unstimulated hHeps (n = 10) were shown to express HLA class I and Fas at 22.1 ± 3.8% and 46.8 ± 8.3%, respectively (mean ± SEM) (Fig. 2). These cells did not express HLA-DR or the costimulatory molecules CD80 and CD40 (Fig. 2A). Positive controls for this staining are shown in Figure 2C. A similar expression pattern was observed in cryopreserved/thawed hHeps and ADHLPCs (Fig. 2A, B). The expression of these cell surface molecules was not modulated in the presence of proinflammatory cytokines (data not shown). Finally, we evaluated the impact of the cell isolation procedure on the hHep immunophenotype by performing immunohistochemistry on liver biopsies from healthy adults. As shown in Figure 2D, hepatocytes in liver tissues present the same profile as isolated hHeps, characterized by an expression of HLA class I and Fas and a negative staining for HLA class II.

Characterization of the immunophenotype of liver-derived cells. Cell surface markers on human hepatocytes, either freshly isolated (hHep), or cryopreserved/thawed (hHepcryo), and adult-derived human liver progenitor cells (ADHLPC) were evaluated by flow cytometry. Isotype controls were performed on all three cell types with similar results. To avoid overcrowding the figure, only one representative isotype control is shown for each staining. (A) Gates were placed based on the corresponding isotype. Results in percent of positive cells are expressed as mean ± SEM. One representative experiment out of eight is shown. (B) Expression of human leukocyte antigen (HLA) class I and FAS (Fas cell surface death receptor; cluster of differentiation 95; CD95) on liver-derived cells. Results in percentage of positive cells are expressed as mean ± SEM. (C) Positive controls for HLA I [peripheral blood mononuclear cells (PBMCs)], HLA II [monocyte-derived dendritic cells (MoDCs)], CD40 [lymphoblastic cell line (LCL)], CD80 (LCL), and FAS [phytohemagglutinin (PHA)-stimulated PBMCs]. (D) Immunohistochemistry was performed on liver section from healthy liver biopsy showing a hepatocyte expression of HLA class I and Fas but not of HLA class II. A positive staining of HLA class II was observed on sinusoidal endothelial cells. Controls were performed by omitting the primary antibody and are shown in the unstained section. One representative experiment out of three is shown. Picture magnification is 200×.
Human Hepatocytes and Liver Progenitor Cells Do Not Induce T-Cell Activation and Proliferation
In order to investigate in vitro the immune response induced by hHeps or ADHLPCs, cells were cocultured with allogeneic PBMCs. We first analyzed the proliferation of allogeneic PBMCs following a 7-day incubation with hHeps or ADHLPCs at various ratios (1:0.5, 1:0.2, 1:0.1). As demonstrated in Figure 3A, no proliferation of PBMCs was detected using the [3H]thymidine incorporation method. The absence of proliferation was confirmed in both the CD4+ and the CD8+ PBMC populations using CFSE staining (data not shown). These results correlated with an absence of upregulation of the intracellular CD40 ligand expression detected on CD4+ T-cells within 24 h of coculture (Fig. 3B). Moreover, after 24 to 96 h of coculture, CD25, CD69, CTLA-4 expression was not upregulated on CD4+ T-cells (Fig. 3C). Finally, no production of IFN-γ was detected in CD4+, CD8+ T-cells, and in NK cells after intracellular staining, using flow cytometry (data not shown). These results demonstrated the absence of early T-cell activation and PBMC proliferation induced by hHeps or ADHLPCs.
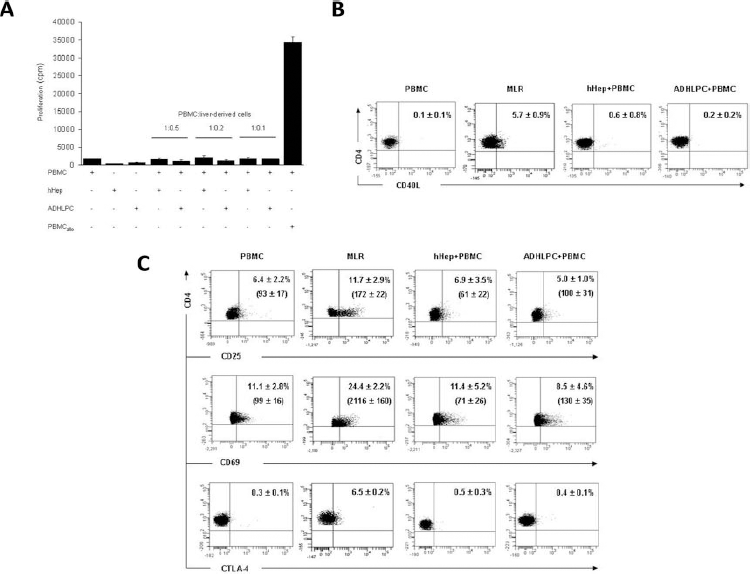
Peripheral blood mononuclear cell (PBMC) proliferation and activation following coculture with liver-derived cells. (A) Allogeneic PBMCs were cocultured with human hepatocytes (hHeps) or adult-derived human liver progenitor cells (ADHLPCs) at varying ratios (1:0.5; 1:0.2; 1:0.1). After 7 days of coculture, the PBMC proliferation was evaluated by [3H] thymidine incorporation. Cells cultured alone were used as negative controls, and PBMCs cultured with allogeneic PBMCs (PBMC) in a mixed lymphocyte reaction (MLR) were used as a positive control. (B) After 24 h of coculture at the ratio 1:0.5, the intracellular expression of CD40 ligand (CD40L) was evaluated on CD4+ T-cells by flow cytometry. (C) After 24 to 96 h of coculture at the ratio 1:0.5, the expression of activation markers CD25, CD69, and cytotoxic T lymphocyte antigen 4 (CTLA-4) was analyzed on CD4+ T-cells by flow cytometry. Results in percentage of positive cells or in median of fluorescence intensity (MFI) are expressed as the mean ± SEM. One representative experiment (at 24 h) out of eight is shown.
Human Hepatocytes Promote a Cell Contact-Dependent Production of IL-10 by Allogeneic Myeloid DCs (mDCs)
Isolated hHeps and ADHLPCs were cultured alone or with allogeneic PBMCs for up to 96 h, and the production of pro- or anti-inflammatory cytokines was evaluated in the culture supernatants using ELISA. When hHeps or ADHLPCs were cocultured with allogeneic PBMCs, levels of IL-6, IFN-γ, and TNF-α were slightly but not significantly increased for up to 96 h compared to cells cultured alone (data not shown). Interestingly, in these settings, hHeps induced a statistically significant increase in IL-10 levels already observed after 24 h of coculture and maintained for up to 96 h, whereas ADHLPCs did not (Fig. 4A). Moreover, no correlation was observed between the presence of a CD45+ contaminating subset in few hHep suspensions and the production of IL-10 in the coculture supernatants. The cellular source that produced IL-10 following coculture with hHeps was identified by flow cytometry. After intracellular staining on the PBMC subsets, mDCs were shown to produce IL-10 after 24 h and up to 96 h (Fig. 4B), whereas CD4+ and CD8+ T-cells, NK, and CD19+ cells did not (data not shown). The phenotype of the IL-10-producing mDCs was also analyzed by flow cytometry. These cells were shown to present an incomplete maturation state characterized by a low expression of HLA-DR but a high expression of CD40 (Fig. 4C). Complementary Transwell experiments were performed in order to rule out a potential role for soluble factors in the IL-10 production. Under such conditions, a very low level of IL-10 was detected in the cocultures by ELISA (Fig. 4A) and intracellular staining after 24 h and up to 96 h (Fig. 4B). These results suggest that hHeps promote the production of IL-10 by allogeneic mDCs in a cell contact-dependent manner, while progenitor cells do not. In parallel, no upregulation of CD40 was observed on mDCs in the cocultures performed in the Transwell chambers (Fig. 4D).
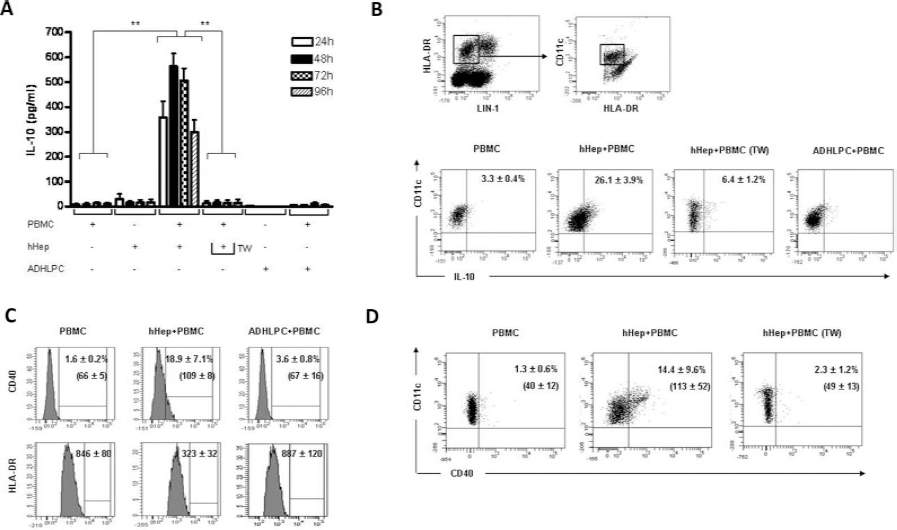
IL-10 producing dendritic cells (DCs) following coculture with liver-derived cells. Allogeneic peripheral blood mononuclear cells (PBMCs) were cocultured with human hepatocytes (hHeps) or adult-derived human liver progenitor cells (ADHLPCs) at the ratio 1:0.5 for 24 to 96 h with or without separation by a Transwell chamber (TW). (A) IL-10 levels in the culture supernatants were determined by ELISA. Cells cultured alone were used as negative controls. Results expressed in pg/ml represent the mean± SD of four independent experiments. **p <0.01. (B) The intracellular production of IL-10 was evaluated in myeloid DCs (mDCs) by flow cytometry. The mDC subset was identified based on the following staining: lineage cocktail (LIN-l)-negative, HLA-DR, and CDllc-positive cells. PBMCs cultured alone were used as negative controls. (C) IL-10-producing mDCs were analyzed for the expression of CD40 and HLA-DR by flow cytometry. (D) The CD40 expression on mDCs after coculture with TW chamber was evaluated by flow cytometry. Results are expressed as the mean ± SD of positive cells (in percent) or MFI. One representative experiment out of three is shown.
hHep-Primed MoDCs Promote Naive CD4+ T-Cell Hyporesponsiveness Through IL-10 Production
To further characterize the consequence of this IL-10 production by hHep-primed MoDCs, MoDCs were cocultured for 48 h with allogeneic hHeps then harvested and cocultured for 7 days with allogeneic naive CD4+ T-cells at various ratios in the presence or absence of a blocking antibody directed against IL-10. The resulting proliferative response was assessed using the [3H]thymidine incorporation method. As expected, MoDCs initially cultured alone triggered the proliferation of naive CD4+ T-cells. Interestingly, ADHLPC-primed MoDCs induced similar levels of naive CD4+ T-cell proliferation as plain MoDCs, while MoDCs previously cocultured with hHeps were shown to induce hyporesponsiveness in naive CD4+ T-cells (Fig. 5A). Moreover, the proliferation of these hyporesponsive naive CD4+ T-cells was partially restored by blocking IL-10, demonstrating that IL-10 contributes to the naive CD4+ T-cell hyporesponsiveness (Fig. 5B). Following coculture with naive CD4+ T-cells and hHep-primed MoDCs, we were not able to detect any expansion of the regulatory T-cell (Treg) population, as Foxp3 expression on CD4+ T-cells was not upregulated following intracellular staining (data not shown).

Analysis of the naive CD4+ T-cell proliferation induced by liver-derived cell-primed monocyte-derived dendritic cells (MoDCs). MoDCs, human hepatocyte (hHep)-primed MoDCs or adult-derived human liver progenitor cell (ADHLPC)-primed MoDCs were cocultured for 7 days with allogeneic naive CD4+ T-cells at varying ratios (0.1:1, 0.02:1, 0.01:1). (A) The proliferation of naive CD4+ T-cells was evaluated by [3H]thymidine incorporation. Cells cultured alone were used as negative controls. The highest proliferation obtained in the coculture of naive CD4+ T-cells and MoDCs was arbitrarily assigned a value of 100% and used as a reference to calculate the percentage of proliferation obtained with the other conditions. (B) Allogeneic naive CD4+ T-cells were cocultured with hHep-primed MoDCs at the ratio 1:0.1 in the presence or absence of anti-IL-10 or isotype control antibodies. Results are expressed as the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
Finally, to determine whether this CD4+ T-cell hyporesponsiveness was antigen dependent or not, CD4+ T-cells from the primary coculture were restimulated for 7 days by MoDCs from the same donor as the primary coculture or from an unrelated donor. CD4+ T-cells were less responsive to restimulation when first stimulated by hHep-primed MoDCs than MoDCs previously cultured in medium alone, regardless of the stimulating MoDC donor used for the restimulation (Fig. 6), suggesting an antigen-independent phenomenon.

Analysis of the CD4+T-cell proliferation after restimulation. CD4+ T-cells previously cocultured with monocyte-derived dendritic cells (CD4MoDC) or CD4+ T-cells previously cocultured with human hepatocyte-primed monocyte-derived dendritic cells (CD4hHep-primed MoDC) were restimulated by monocyte-derived dendritic cells from the same donor as the primary coculture (MoDC) or from an unrelated donor (MoDCun) at the ratio CD4+ T-cells:dendritic cells 1:0.1. After 7 days, the CD4+ T-cell proliferation was evaluated by [3H]thymidine incorporation. Cells cultured alone were used as negative controls. Results are expressed as the mean ± SD of three independent experiments. **p < 0.01.
The Differentiation of Liver Progenitor Cells in Hepatocyte-Like Cells Does Not Modify Their Immunological Profile
Finally, to assess whether differentiation in hepatocyte-like cells may influence the immunogenicity, all the experiments were performed in the same conditions after in vitro hepatogenic differentiation of ADHLPCs. All results were comparable to undifferentiated ADHLPCs as no immune response was detectable (data not shown).
Discussion
In the context of LCT, where the infused cells come into direct contact with recipient immune cells following intraportal injection, it was important to investigate the propensity of these cells to induce an immune response. Indeed, it is not yet entirely clear whether the short-lived efficacy of LCT is due to progressive loss of cell function or to an eventual clearance of the cells by the immune system of the host. In addition, immunogenicity data could shed some light on the necessity to subject LCT patients to a full immunosuppressive regimen. In this context, we demonstrated that hHeps and ADHLPCs presented a low immunogenic phenotype and did not induce either early T-cell activation or PBMC proliferation in vitro. Following coculture with allogeneic hHeps, we observed the emergence of IL-10 producing mDCs characterized by a partial maturation state and an ability to promote a cell contact- and IL-10-dependent hyporesponsiveness of naive CD4+ T-cells, even after restimulation. On the contrary, such immune response was not elicited by undifferentiated or differentiated liver progenitor liver cells, which did not seem to induce any detectable immune reaction in vitro.
The immunological phenotype of hHeps and ADHLPCs is characterized by a moderate expression of HLA class I and no expression of HLA class II and the costimulatory molecules CD80 or CD40. These observations correlated with previous studies demonstrating the expression of HLA class I on human hepatocytes with an expression of HLA class II restricted to hepatocytes from some diseased livers (21,27). A similar expression pattern was also observed on murine hepatocytes (6,14). The modulation of HLA class I and II expression on human and rodent hepatocytes by inflammatory cytokines is still debated (8,14,24,39). In our hands, no modification of hHeps' immunophenotype was observed with graded doses of inflammatory cytokines susceptible to be involved in graft rejection (IFN-γ and TNF-α). The Fas—Fas ligand interaction is an important mechanism involved in the hepatocyte allograft rejection (25,29,34). Indeed, the surface expression of Fas on donor cells may expose them to apoptotic cell death induced by the host-activated T-cell expressing Fas ligand molecules. Moreover, the downregulation of Fas expression on rodent hepatocytes has been associated with prolonged cell graft acceptance (29). Here we confirmed that hHeps and ADHLPCs commonly express Fas, which does not procure them the same immunological privilege as described in the eyes or the testis (16).
The effector mechanisms involved in the immune response following transplantation are complex, somehow redundant, and dependent on the origin of the transplanted tissue (1). In our study, to better mimic and monitor the immune response in vitro, allogeneic PBMCs were used as responder cells in a coculture model with hHeps or ADHLPCs. A similar coculture model in which PBMCs were stimulated by a donor-specific alloantigen was previously proposed to measure the T-cell alloreactivity in the context of liver transplantation (37). While hHeps are directly infused in the portal vein of the recipient, this model was preferred to a whole blood model in order to avoid the tissue factor-dependent procoagulant activity of hHeps and the inflammatory cascade (46). Under such conditions, hHeps and ADHLPCs were unable to induce any proliferation of CD4+ and CD8+ responder T-cells. This absence of T-cell proliferation was correlated with an absence of early T-cell activation markers. Furthermore, the production of other proinflammatory cytokines (IL-6, TNF-α), evaluated in the supernatant of the cocultures with hHeps or ADHLPCs, was not significantly increased. Taken together, these data demonstrated the low immunogenicity of these liver-derived cells, which is consistent with the well-known immunoprivi-leged status of the liver. But this observation might appear as a paradox since in vivo studies in mice showed allogeneic murine hepatocyte rejection by novel and specific patterns of immune responses implicating both CD4+ and CD8+ T-cells with a plausible role for host APCs (20). This discrepancy could be due to the fact that in a murine model, the allogeneic isolated hepatocytes were mostly infused into the spleen, a method that seems to be more immunogenic since murine liver DCs are stronger activators of immunity than murine liver DCs (35). Moreover, our study was carried out on isolated human cells using in vitro experiments performed under clinical conditions. To this day, very few data are available on the immunogenicity of isolated hHeps. One study led by Allen et al. has suggested a T-cell-mediated rejection of allogeneic hHeps following liver cell transplantation performed in one patient with Crigler Najjar disease (2). It has to be noted that in this article, the CD8+T-cell alloreactivity was evaluated by restimulating recipient PBMCs with irradiated PBMCs or transfected B-lymphoblastoid cell lines (B-LCL) presenting some HLA antigens in common with the donor hepatocytes. This could have an impact on the results because PBMCs and B-LCL are of immune origin and therefore present a different immunogenic potential. In addition, unlike hHeps, these cell types do not reflect the liver environment.
The contribution of IL-10 to the prevention of liver graft rejection has already been suggested by detection of this cytokine in the serum of pediatric recipients with liver graft acceptance (13). In addition, a highly significant correlation has been shown between a stable liver graft function and the production of IL-10 by PBMCs of liver transplanted recipients after in vitro stimulation by donor-specific alloantigens (19). In our experiments, we observed a significant cell contact-dependent production of IL-10 by the mDC subset following coculture with hHeps. Although IL-10 could be induced by various cell types, we found no correlation between the production of IL-10 and the presence of a CD45+ subset contaminating some of the hHep suspensions.
The essential role of DC subsets in the modulation of the T-cell response has been extensively described in the literature (23,31). In this study, we demonstrated that hHeps have the capacity to modulate the DCs, thereby polarizing the T-cell response, as demonstrated by the hyporesponsiveness of the naive CD4+ T-cell after restimulation by hHep-primed MoDCs. The involvement of IL-10 was demonstrated by the ability of anti-IL-10 antibodies to block hHep-primed MoDC-induced CD4+ T-cell hyporesponsiveness. Although we did not have enough cells to determine the impact of anti-IL-10 antibodies on the proliferation of CD4+ T-cells stimulated with naive MoDCs, we are pretty confident that the effect depicted in Figure 5A is specific as several researchers have previously demonstrated that anti-IL-10 alone does not induce CD4+ T-cell proliferation (15,48). In a similar in vitro model, Cabilic et al. reported that human MoDCs generated in a hepatic environment or cocultured with rat liver cells present the capacity to produce IL-10, leading to T helper2 cell (Th2) polarization (10). Furthermore, a recent study showed that human DCs isolated from liver were poorly immunogenic and promoted T-cell hypo-responsiveness and Treg induction through an IL-10-dependent mechanism (3). Moreover, the generation of Foxp3+ Tregs from naive CD4+ T-cells using MoDCs was also described (43). In our study, we did not find any evidence for the generation of Foxp3+ Tregs by hHep-primed MoDCs. But the capacity of MoDCs to induce the generation of Tregs from allogeneic naive CD4+ T-cells was assessed only after repetitive in vitro restimulation (22). Interestingly, the IL-10-producing mDCs observed in our study presented an incomplete maturation state. Based on the old paradigm, one could argue that these results cannot be reconciled with the induction of tolerance. However, growing evidence indicates that DC maturation per se is not representative of their immunogenicity. Indeed, it has been shown that the induction of tolerance by DCs requires a partial maturation and that the maturation microenvironment plays a key role in the ability of DCs to induce tolerance versus immunity (28,38).
Beyth et al. have reported the ability of human bone marrow-derived mesenchymal stem cells (MSCs) to inhibit T-cell responses by the induction of regulatory APCs (5). In our study, ADHLPCs were not able to induce the generation of IL-10-producing mDCs with tolerogenic function, even after in vitro differentiation, which could suggest an incomplete hepatogenic differentiation (26). However, the immunosuppressive activity of human MSCs can be mediated by several distinct mechanisms (33). Therefore, further investigations are needed to better explore the potential immunosuppressive properties of ADHLPCs.
In conclusion, both human hepatocytes and liver progenitor cells were associated with a low immunogenic profile. In addition, the data obtained using isolated hepatocytes support the concept that the liver environment can promote the induction of tolerogenic DCs. These observations could have important implications in better understanding hepatic immunity in LCT and defining new strategies for immunosuppressive regimens.
Footnotes
Acknowledgments
Liver cell transplantation program was supported by Direction Générale des Technologies, de la Rechercheet de l'Energie, Région Wallonne (WALEO/HEPATERA). This work was supported by a grant (FRIA FC 75428) from Fonds de la Recherche Scientifique-FNRS and by a grant from the government of the Walloon Region (FIRST 051/6261-CRISTALL study). We also thank the Fondation Saint-Luc for continuous support. The authors declare no conflicts of interest.