
Abstract
Human umbilical cord blood (CB)-derived unrestricted somatic stem cells (USSCs) have previously been demonstrated to have a broad differentiation potential and regenerative beneficial effects when administered in animal models of multiple degenerative diseases. Here we demonstrated that USSCs could be induced to express genes that hallmark keratinocyte differentiation. We also demonstrated that USSCs express type VII collagen (C7), a protein that is absent or defective in patients with an inherited skin disease, recessive dystrophic epidermolysis bullosa (RDEB). In mice with full-thickness excisional wounds, a single intradermal injection of USSCs at a 1-cm distance to the wound edge resulted in significantly accelerated wound healing. USSC-treated wounds displayed a higher density of CD31+ cells, and the wounds healed with a significant increase in skin appendages. These beneficial effects were demonstrated without apparent differentiation of the injected USSCs into keratinocytes or endothelial cells. In vivo bioluminescent imaging (BLI) revealed specific migration of USSCs modified with a luciferase reporter gene, from a distant intradermal injection site to the wound, as well as following systemic injection of USSCs. These data suggest that CB-derived USSCs could significantly contribute to wound repair and be potentially used in cell therapy for patients with RDEB.
Introduction
There has been a growing interest in utilizing bone marrow (BM) or cord blood (CB) and its derived stem cells for the treatment of degenerative diseases, such as myocardial infarction and neurological disorders (24). Preclinical studies also indicated that stem cells in the BM or CB contribute to wound healing and may rescue a group of basement membrane defects, epidermolysis bullosa (EB) (9,18,34). The most severe form, recessive dystrophic EB (RDEB), is caused by mutations in the COL7A1 gene that encodes type VII collagen (C7) (13). The C7 protein is synthesized by both basal keratinocytes and dermal fibroblasts and is a major component in the anchoring fibrils at the epidermal–dermal junction (18,36). Patients with RDEB suffer from blistering, repeated wounding and healing in the skin, oral mucosa, and gastrointestinal/genitourinary tracts (7,8).
A recent clinical trial supported the beneficial effects of allogeneic stem cell transplantation in patients with RDEB. A majority of recipients showed increased C7 protein deposition at the dermal–epidermal junction and exhibited clinical improvement (37). However, there was no distinct anchoring fibril formation in the recipient skin. Donor cell chimerism has been observed in the recipients' skin, including not only cluster of differentiation 45 positive (CD45+) hematopoietic cells but also nonhematopoietic, nonendothelial donor cells in the vicinity of the epidermal–dermal junction (33). Such nonhematopoietic, nonendothelial cells may have played an important role in the regeneration of RDEB skin; however, their identities remain to be determined.
BM and CB are rich sources for nonhematopoietic stem cells and progenitors with regenerative potential (24). One such population, multipotent mesenchymal stem cells (MSCs), demonstrated the ability to promote dermal regeneration and wound healing (2,23,26,29,31,39). This has made MSCs an attractive treatment option in wound healing applications, especially to resolve hard-to-heal wounds or ulcers (16). Meanwhile, MSCs express C7 protein and alleviated the RDEB phenotype both in an RDEB animal model and in human subjects (1,5).
In addition to MSCs, human CB contains unrestricted somatic stem cells (USSCs) (21). USSCs are considered a precursor to MSCs and can be distinguished from MSCs by their higher expansion capacity, broader differentiation ability, and differential expression of genes including δ-like 1/preadipocyte factor 1 (DLK1) and the homeobox (HOX) gene clusters (19,22,25). USSCs have the potential to differentiate in vitro to osteoblasts, chondrocytes, and hematopoietic and neuronal cells and in vivo to bone, cartilage, hepatocytes, hematopoietic cells, myocytes, etc. USSCs constitutively express a series of cytokines including stem cell factor, leukemia inhibitor factor, vascular endothelial growth factor (VEGF), stromal cell-derived factor (SDF) 1, etc., and have strong hematopoietic stimulating activity (20). Similar to MSCs, USSCs lack expression of immunorelevant adhesion and costimulatory molecules (38). However, immunosuppression by USSCs is conditional and dependent on tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) (38). Administration of USSCs in multiple animal disease models has resulted in the promotion of bone healing and recovery from neural injury and myocardial infarction (10,11,17,35).
In this study, we demonstrated that USSCs express C7 protein and have the ability to undergo in vitro differentiation to keratinocyte-like cells. We further demonstrated that USSCs promote wound healing and skin regeneration in a murine wounding model. In vivo bioluminescence imaging (BLI) provided the first evidence for the migration of a subset of not only systemically but also locally injected USSCs to skin wounds. Our results support the therapeutic potential of CB-derived USSCs in the treatment of RDEB.
Materials and Methods
Isolation of USSCs
Using CB collection kits from Pall Medical (East Hills, NY, USA), CB was collected on site at New York Presbyterian Morgan Stanley Children's Hospital, Columbia University Medical Center, following the institutional review board (IRB) approval. Individual informed consent was waived per IRB approval. No donor data were collected. CB was stored at room temperature (~25°C) and processed within 2 days postcollection. Each sample was processed independently as described. CB was first diluted 1:1 with phosphate-buffered saline (PBS; Hyclone, Logan, UT, USA), and mononuclear fraction was obtained from the buffy following Ficoll-Paque™ PLUS (GE Healthcare™ PLUS, Uppsala, Sweden) gradient separation. The mononuclear cells were washed twice again with PBS and plated at 5 × 106 cells/ml in tissue culture flasks in USSC initiation medium following a published method (21). The USSC initiation medium is composed of 69% DMEM low glucose (GIBCO, Auckland, New Zealand), 30% fetal bovine serum (FBS; HyClone), 1% penicillin– streptomycin solution, 10−7 M of dexamethasone (Sigma, St. Louis, MO, USA), and 2 mM of UltraGlutamine (Lonza, Walkersville, MD, USA). Half of the initiation medium was changed at 24 h, 1 week, 2 weeks, and 3 weeks or until USSC colonies were observed. After the appearance of USSC colonies, cells were expanded in the same medium without dexamethasone.
Flow Cytometry
USSCs were immunophenotyped by the following antibodies: fluorescein isothiocyanate (FITC)-conjugated Ab CD90, CD14, CD31, CD50, CD106, CD45, CD34, stage-specific embryonic antigen (SSEA)-4, and isotope control (BD Biosciences, San Jose, CA, USA), phycoerythrin (PE)-conjugated CD146, CD73, platelet-derived growth factor receptor (PDGFR) a, isotope control (BD Biosciences), and VEGF receptor 2/kinase insert domain receptor (VEGF-R2/KDR; R&D systems, Minneapolis, MN, USA), StainAlive TRA-1-60 DyLight-488 (Stemgent, Cambridge, MA, USA) and unconjugated Ab CD105, CD144, CD33, isotope control (BD Biosciences), CD49e [integrin a 5 (INTa5)], chemokine C-C motif receptor 1 (CCR1), CCR2, CCR7, chemokine C-X-C motif receptor 4 (CXCR4), and CD29 (INTβ1; AbCam, Cambridge, MA, USA). One hundred-microliter cell aliquots, each containing 5 × 105 cells, were resuspended in ice-cold PBS containing 10% FBS and 1% sodium azide (Sigma). Antibodies were added at a dilution according to the manufacturer's recommendation, respectively, and incubated on ice for 30 min in the dark. If unconjugated primary antibodies were used, the cells were washed with ice-cold PBS three times by centrifugation and resuspended in PBS containing 3% bovine serum albumin (BSA; Miltenyi Biotec, Cambridge, MA, USA). Fluorochrome-labeled secondary antibody (Invitrogen/Molecular Probes, Eugene, OR, USA) was then added and incubated on ice for 30 min in the dark. After washing with PBS, the samples were analyzed by flow cytometry using Cell Quest Software (BD Biosciences).
RT-PCR and qPCR
RNA extraction was performed with an RNeasy kit (Qiagen, Valencia, CA, USA). cDNA was obtained through Clontech Advantage RT-for-PCR kit (Mountain View, CA, USA), and PCR was done under standard PCR conditions (94°C for 30 s, 60°C for 30 s, 68°C for 45 s, for 30 cycles) with Titanium™ DNA Taq Polymerase from Clontech. Primer sequences are listed in Table 1. For qPCR, the reactions were performed using SYBR-Green PCR master mix (Applied Biosystems, Warrington, UK) in a 7300 Real-Time PCR System (Applied Biosystems). As an internal control, levels of glyceraldehyde-phosphate dehydrogenase (GAPDH) were quantified in parallel with target genes. Normalization and fold changes were calculated using the ΔΔCt method. The control human male fibroblasts and keratinocytes were kindly provided by the Skin Disease Research Center (SDRC) of Columbia University. The female H9 embryonic stem (ES) cells were kindly provided by Dr. GordanaVunjak-Novakovic at Columbia University. The human acute lymphoblastic leukemia cell line from a female, RS4;11 (ATCC, Manassas, VA, USA) was used as a negative control for the COL7A1 expression. To analyze the effects of 5-azacytidine on pluripotent gene expression, exponentially growing USSCs and fibroblasts in their growth medium were also treated with 3 μM 5-azacytidine (Sigma) for 8 h, before extraction of their RNA.
List of Primers

OCT4, octamer-binding transcription factor 4; Col7A1, collagen type VII; KRT14, keratin 14; TAP63, tumor protein 63; DSG3, desmoglein 3; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; DLK, δ-like 1/preadipocyte factor 1
Bisulfite DNA Analysis
DNA bisulfite conversion was conducted from human ES, fibroblasts, and USSCs, respectively, using EZ DNA Methylation-Direct™ Kit (Zymo Research Corp., Orange, CA, USA). The regions of interest (ROI) were amplified by PCR using bisulfite primers, as listed in Table 1. The amplified PCR products were purified using QIAquick PCR purification Kit (Qiagen) and cloned into pGEM-T Easy Vector (Promega, Madison, WI, USA) followed by transformation into high-efficiency JM109 competent cells (Promega). Colony PCR was performed from the transformed colonies using SP6 and T7 primers, and DNA sequencing was performed by Genewiz, Inc. (South Plainfield, NJ, USA).
Alkaline Phosphatase and Immunofluorescence Staining
For AP staining, USSCs were fixed with 4% paraformaldehyde (PFA; Electron Microscopy Services, Hatfield, PA, USA)/PBS and washed with AP staining buffer [100 mM Tris (pH 9.5), 50 mM MgCl2, 10 mM NaCl, and 1% Tween 20 in H2O; all Sigma]. The plate was then incubated with 5-bromo-4-chloro-3-indolyl phosphate and 4-nitro blue tetrazolium chloride (Roche Applied Science, Indianapolis, IN, USA) in AP staining buffer for the color development. The percentage of AP-positive cells was calculated based on the number of AP-positive cells and the total number of cells in three random fields of sparsely plated USSCs. Experiments were repeated in three independently derived USSC lines. For immunofluorescent staining, the cell culture or the cryosection of skin biopsies was fixed with 4% PFA/PBS. After 1 h of blocking using 10% goat or donkey sera (Life Technologies, Grand Island, NY, USA) in 0.1% Triton X-100 (Sigma)/PBS, the samples were incubated for 1 h at room temperature or overnight at 4°C with primary antibodies against keratin 14 (KRT14; Covance, Princeton, NJ, USA), COL7A1 (C7) (LH7.2; Santa Cruz Biotechnology, CA, USA), CD31 (Abcam), and INTα5 (Abcam). After three rinses with PBS, incubation with appropriate secondary antibodies (Molecular Probes, Eugene, OR, USA) was performed for 1 h at room temperature. Nuclei staining was performed by Vectashield® mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA).
Keratinocyte Differentiation
To induce keratinocyte differentiation, USSCs were seeded in 0.1% gelatin (Millipore)-coated plates and incubated in the defined keratinocyte serum-free medium (K-SFM; Invitrogen, Carlsbad, CA, USA) supplemented with 10 ng/ml bone morphogenetic protein 4 (BMP4; Invitrogen), 0.3 mM ascorbic acid (Sigma), and 3 ng/ml recombinant epidermal growth factor (EGF; Invitrogen). The medium was changed every other day for 7 days. The defined K-SFM medium without the above supplements was then used for continued culturing.
Wound Healing Model
A 1 cm × 1 cm full-thickness excision wound was created on the midback of each 6- to 10-week-old immunocompromised mouse (NOD.CB17-Prkdcscid/J, Jackson Laboratories, Bar Harbor, ME, USA) after hair removal using Veet gel cream (Reckitt Benckiser Inc., Parsipanny, NJ, USA) and anesthesia (Isoflurane; Phoenix Pharmaceutical Inc., St. Joseph, MO, USA). The wound was then covered by Tegaderm™ film (Express Medical Supply Inc., St. Paul, MN, USA) to prevent desiccation. Four hours following induced wounding, 1 × 106 USSCs in 100 μl PBS was injected intradermally using a 27-gauge 1/2-inch syringe (Becton Dickinson, Franklin Lakes, NJ, USA) at a single injection site 1 cm away from the midpoint of the right side of the wound. The mice injected with the same volume of PBS were used as negative controls. Digital photographs of wounds were taken each day under anesthesia. The wound area was measured by tracing the wound margin and quantitated using Image J software (NIH, Bethesda, MD, USA). The percentage of wound area was calculated as area of actual wound/area of original wound × 100. Two-way ANOVA analysis followed by Bonferroni post hoc test was used to determine any significant difference between USSC- and PBS-treated groups in the rate of wound closure. A value of p < 0.05 was considered significant. Full-thickness skin biopsies including the wounds and about 0.3-cm flanking unwounded skin were extracted at selected days for histological analysis and immunocytochemistry. The embedding, sectioning, and hematoxylin and eosin (Sigma) analysis were processed following standard procedures at the Skin Disease Research Center (SDRC) of Columbia University. For in vivo imaging, a total of 1 × 106 USSCs were intradermally injected at four sites around the wound, or a total of 1.5 × 106 USSCs were injected via the tail vein 2 h postexcisional wounding. All animal studies were conducted using protocols approved by both the Columbia University and New York Medical College Animal Use Committees.
Labeling of Green Fluorescent Protein (GFP)-Luciferase Reporter Genes in USSCs and In Vivo Bioluminescent Imaging
The lentiviral construct, pSico PolII-eGFP-Luc2, was generously provided by Dr. Glenn Merlino at the National Cancer Institute (6). For the lentiviral production, the 293T cells (ATCC) were transfected with the lentiviral construct mixed with the ViraPower™ packaging mix (Invitrogen), using the FuGENE 6 transfection reagent (Roche). The cell culture supernatant containing the lentiviral particles was collected 48 and 72 h posttransfection and passed through the 0.75-μm syringe filters (Nalgene, Rochester, NY, USA) to remove any floating cells.
To transduce USSCs, the lentiviral supernatant was premixed with 6 μg/ml of polybrene (Sigma) and then added to the USSC culture plate for overnight incubation. Green fluorescent protein (GFP) expression was examined under a fluorescence microscope, and the GFP-positive cells were sorted in the fluorescence-activated cell sorting core facility (Herbert Irving Comprehensive Cancer Center of Columbia University).
The mice used for BLI were of the same strain as those used for the wound healing model. Mice were imaged using a Xenogen IVIS imaging system (Hopkinton, MA, USA), per the manufacturer's directions. Mice were anesthetized with isofluorane (Phoenix Pharmaceutical Inc.) and injected intraperitoneally with 50 mg/kg of XenoLight RediJect D-Luciferin Ultra (Caliper, Hopkinton, MA, USA) 15 min before imaging. The images were taken at 2 h post-USSC injection and selected days afterward. Living Image 4.1 software (Caliper) was used to quantify total photon flux (photons/s/cm2/sr) in ROIs.
Results
Isolation and Characterization of USSCs
USSCs were initiated from CB by plating the mono-nuclear cells in the presence of 30% FBS and 10−7 M of dexamethasone, as previously described in the literature (21). About 50% (n = 26) of CB gave rise to spindle-shaped colonies, with a frequency of 1–10 per CB unit. The colonies were then trypsinized and expanded as monolayer cells. Reverse transcription polymerase chain reaction (RT-PCR) analysis was conducted on derived colonies for the expression of δ-like 1/preadipocyte factor 1 (DLK-1/PREF1), a marker to distinguish USSCs from CB-derived MSCs (19) (Fig. 1A). Only the cells expressing DLK-1 and having been passaged for more than six times were used in the following studies.

Characteristics of USSCs. (A) A representative RT-PCR analysis on the expression of δ-like 1/preadipocyte factor 1 (DLK-1) in cord blood (CB)-derived clones. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. (B) Immunophenotypic analysis of unrestricted somatic stem cells (USSCs) by flow cytometry. USSCs were labeled with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated control isotope IgG (gray peaks) or antibodies against the indicated cell surface proteins (black peaks). CD90, cluster of differentiation 90; PDGFRα, platelet-derived growth factor receptor α; CCR7, chemokine C-C motif receptor 7; CXCR4, chemokine C-X-C motif receptor 4; KDR, kinase insert domain receptor/VEGF receptor 2; SSEA4, stage-specific embryonic antigen 4; (C) Representative alkaline phosphatase (AP) staining with USSCs (top image) and human fibroblasts (hFb) (bottom image). Scale bar: 100 μm. The percentage of AP-positive cells was calculated in sparsely plated USSCs, and the experiment was repeated in three independently derived DLK-1-expressing USSC lines. (D) RT-PCR analysis for the expression of octamer-binding transcription factor 4 (Oct4) and Nanog in USSCs, compared with human ES cells and fibroblasts. USSCs showed a low level of Oct4 and Nanog gene transcription, and treatment with 3 μM DNA methylation inhibitor 5-azacytidine (5-azaC) further enhanced their gene expression in USSCs. All the lanes were set up in the same PCR reaction and loaded on the same gel. This set of experiments was repeated twice with RT-PCR and quantitative RT-PCR, respectively.
The USSC population was negative for expression of CD45, CD34, CD33, CD31, CD144, CD14, CD50, KDR, and CD106 but expressed significantly higher levels of CD90, CD73, CD105, CD146, CD49e/INTα5, and CD29/INTβ1, consistent with a previous report (21). An example of the immunophenotype of USSCs is presented in Figure 1B. About 10.76% and 22.1% of USSCs expressed CCR7 and CXCR4, receptors for chemoattractant proteins secondary lymphoid tissue chemokine/chemokine C-C motif ligand 21 (SLC/CCL21) and SDF1, respectively. The interaction between CCR7/CCL21 was suggested to be the mechanism for the migration of MSCs to the wound, and the interaction between CXCR4/SDF1 mediates the migration of MSCs to the BM (29,40). We also showed that about 86% of USSCs express PDGFRα. It has recently been demonstrated that PDGFRα-positive cells in BM contributed to the regeneration of epidermis after skin grafting in vivo (32).
In addition, 57.46% of USSCs (±10.97%) displayed SSEA-4, usually expressed on the surface of human ES cells, teratocarcinoma stem cells, and embryonic germ cells (Fig. 1B). However, USSCs were negative for the pluripotent stem cell marker TRA-1-60 (Fig. 1B). To further determine the stem cell properties, we performed AP staining and revealed a positive AP activity in about 33% (±4.8%) of the USSCs (Fig. 1C). Moreover, compared with a complete absence of octamer-binding transcription factor 4 (Oct4) and Nanog gene transcription in human fibroblasts, a minimal level of such pluripotency gene expression was detected in USSCs using isoform-specific and intron-spanning primers (Fig. 1D). As it was recently demonstrated that USSCs exhibit an uncommitted epigenetic signature for the major pluripotency genes (28), we treated USSCs and fibroblasts in parallel with DNA methylation inhibitor 5-azacytidine to determine the effects of chromatin modification on the expression of these genes. The RT-PCR and qPCR analysis indicated that the treatment led to about a 10-fold increase in the expression of the genes in USSCs, but not in fibroblasts (Fig. 1D and data not shown). However, the level of Oct4 and Nanog expression in USSCs, even after gene activation, was still low and represents only about 1% and 3% of that in the human ES cells, respectively (data not shown).
Bisulfite sequencing on the upstream regulatory regions of both Oct4 and Nanog genes further revealed an intermediate DNA methylation pattern in USSCs compared with human ES and fibroblasts (Fig. 2A, B). The cytosine-phosphate-guanosines (CpGs) within the Oct4 proximal promoter (PP) were mainly unmethylated in USSCs, consistent with the previous report (28). This element includes the phylogenetically conserved region 1 (CR1) and contains GC-rich specificity protein 1 (Sp1)-like sequence and three hormone response element (HRE) half sites. In comparison, the promoter region that extends up to 1,000 bp upstream was more heavily methylated. An analysis on the more upstream regulatory region revealed that USSCs were mainly un methylated at the sequence that encompasses the CR3 and were partially methylated at the CR4, where the Oct4/sex-determining region Y box 2 (Sox2)-binding domain resides. Similarly, the promoter and enhancer regions of the Nanog gene in USSCs were also partially methylated. The incomplete DNA methylation at the key regulatory regions of Oct4 and Nanog genes is consistent with the minimal expression of these pluripotent master genes and their being readily activated following the treatment of the DNA methylation inhibitor.

Mosaic DNA methylation pattern. Mosaic DNA methylation pattern in USSCs at the core regulatory regions of the Oct4 (A) and Nanog genes (B), compared with human ES cells and fibroblasts. CR, conserved region; DE, distal enhancer; PE, proximal enhancer; PP, proximal promoter.
We also demonstrated by RT-PCR that USSCs express COL7A1 mRNA, similar to human keratinocytes and dermal fibroblasts (Fig. 3A). A leukemia cell line, RS4;11, was used as a negative control for COL7A1 expression. The expression of C7 protein has also been demonstrated following immunostaining on USSCs using human C7 antibody (LH7.2) (Fig. 3B). This key finding supports the therapeutic potential of USSCs in rescuing the defective anchoring fibril formation in patients with RDEB.

USSCs express Col7A1 and can be differentiated into keratinocyte-like cells. (A) RT-PCR analysis indicated that USSCs expressed collagen type VII (Col7A1 or C7) mRNA, similar to human keratinocytes and fibroblasts. A leukemia cell line RS4;11 was used as a negative control for gene expression. Two sets of Col7A1 RT-PCR primers were tested, with the same results. Experiments were repeated three times. (B) Immunostaining of USSCs with human C7 antibody (LH7.2) (green). Nuclei were stained by 4′,6-diamidino-2-phenylindole (DAPI; blue). (C) Directed differentiation of USSCs into keratinocyte-like cells in vitro. RT-PCR analysis showed that the differentiated USSCs express keratinocyte markers including keratin 14 (KRT14), keratin 5 (KRT5), tumor protein 63 (TAP63), and DSG3. These markers were negative in undifferentiated USSCs and positive in human keratinocytes. GAPDH was used as an internal control. All the lanes were set up in the same PCR reaction and loaded on the same gel. The differentiation experiment was repeated three times with similar results. (D) Immunostaining of human KRT14 (K14; red) in differentiated USSCs. Nuclei were stained by DAPI (blue). Scale bar: 10 μm.
In Vitro Differentiation of USSCs to Keratinocyte-Like Cells
A recent study demonstrated the ability of USSCs to differentiate along the ectoderm lineage into neurons (11). We also demonstrated that USSCs were able to differentiate into neural stem cells that later gave rise to neurons, oligodendrocytes, and astrocytes (data not shown). Here we investigated whether USSCs are able to differentiate into keratinocytes. We first applied a combination of retinoic acid and BMP4 that induced differentiation of human-induced pluripotent stem (iPS) cells into keratinocytes (15). However, under such experimental conditions, we did not detect any keratinocyte-specific gene expression in USSCs. We then induced USSCs in defined K-SFM supplemented with BMP4, ascorbic acid, and EGF2 for 7 days, followed by continued culturing in K-SFM (29). USSCs started to change their morphology from elongated spindle shape to a rounded/polygonal shape within 2 weeks of induction. RT-PCR analysis on the 30-day culture indicated that the cells acquired the expression of KRT14, KRT5, tumor protein 63 [(TA)P63], and desmoglein 3 (DSG3) (Fig. 3C). The expression of these genes is a hallmark of keratinocyte differentiation, and their expression was not detected in uninduced USSCs. The expression of KRT14 was also demonstrated in induced USSCs by immunostaining (Fig. 3D). This suggests the ability of USSCs to differentiate into keratinocyte-like cells under defined in vitro conditions. However, it was also noted that the expression of the genes, especially KRT5, P63, and DSG3, is at a much lower level in the USSC differentiation culture conditions than that in human keratinocytes. This suggests that either the differentiation was not complete or only a small subset of USSCs was able to differentiate into keratinocytes.
Intradermal Injection of USSCs Promoted Wound Healing
An important phenotype in the skin of RDEB patients is continuous blistering and wounding. Therefore, our initial approach to investigate the therapeutic potential of USSCs for patients with RDEB was to determine their ability to alleviate wounding in immunocompromised mice with 1 × 1-cm2 full-thickness excisional wounds. Four hours postwounding, 1 million USSCs resuspended in 100-μl PBS were intradermally injected at a single site at a 1-cm distance from the margin of the wound. The same volume of PBS was injected in parallel and served as a negative control. Figure 4A shows representative USSC and PBS-treated wounds on days 1, 2, 7, 10, and 15 postwounding. The wound size was measured in each group daily for 15 days and analyzed by two-way ANOVA followed by Bonferroni post hoc test. The USSC-treated wounds showed an accelerated healing compared with the PBS-treated control, and a significant difference [F(1, 168) = 50.8, p < 0.01] was observed on days 6–10 postwounding (Fig. 4B).
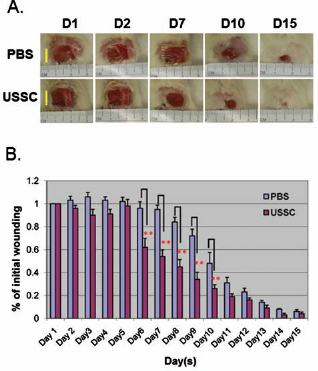
USSC promoted wound healing in a murine model with 1-cm2 full-thickness excisional wound. (A) Representative USSC and phosphate-buffered saline (PBS; vehicle)-treated wounds on days 1, 2, 7, 10, and 15 postwounding. (B) Two-way ANOVA on the wound size, followed by Bonferroni post hoc test showed a significant difference [F(1, 168) = 50.8, p < 0.01] on wound healing on days 6, 7, 8, 9, and 10. Data are presented as mean ± SEM. n = 8~9/treatment. **Statistically significant difference between PBS and USSC treatment at 1% level. Scale bar: 1 cm.
Histological analysis on skin samples taken at different time points indicated that USSCs promoted epithelialization and facilitated formation and remodeling of epidermis in the wound (Fig. 5). On day 3 postwounding, hematoxylin and eosin analysis indicated that there was an enhanced cellularity in the wounds of the USSC-treated mice than in the PBS control (shown by the arrow). On day 8, reepithelialization of the wound area was evident in the USSC-treated group, in contrast to the control group that showed ulcerated wounds with fibrinous debri (Fig. 5). On day 16, wounds were mostly closed in both the USSC- and PBS-treated groups, and there was no apparent difference as determined by the visual appearance of the wounded area. However, upon microscopic analysis, the PBS-treated wounded area showed a disorganized epidermal structure (Fig. 5). In comparison, USSC-treated wounds exhibited a distinct epidermis with multiple layers of keratinocytes, as determined by their positive KRT14 staining (data not shown). Moreover, USSC-treated wounds also showed an early appearance of skin appendages at the center of the wound, which also stained positive for KRT14 (Fig. 5 and data not shown). On day 40, both USSC- and PBS-treated wounds demonstrated a thin epidermal layer, suggesting that they had undergone skin remodeling (Fig. 5). However, the PBS-treated wounds healed with more fibrotic tissue and with an absence of epidermal appendages. In significant contrast, multiple skin appendages, that is, sebaceous glands and hair follicles, were present in the wound center of the USSC-treated skin, as determined by their morphology, positive K14 staining, and presence of hair shafts (Figs. 5 and 6A). Similar skin appendages were observed in the wound sections in six out of nine mice analyzed between day 26 and day 44 post-USSC injection (data not shown).

Hematoxylin and eosin staining of skin biopsies from mice treated with PBS and USSCs. On day 3, USSC-treated wound showed a higher level of cellularity compared with the PBS-treated group, especially underneath the surface of the wound (indicated by arrow). On day 8, the protrusion of epidermis became apparent in the USSC-treated wound. On day 16, USSC-treated mice showed the presence of multilayer epidermis and an initial appearance of gland (inset). On day 40, USSC-treated wounded area showed less fibrosis compared with the PBS group and exhibited clear presence of multiple skin appendages. The insets in the day 40 group magnified the formation of fibrotic tissue (following PBS injection) and sebaceous gland (following USSC treatment). Scale bar: 50 μm.
USSCs Promoted Wound Healing Via a Paracrine Effect
To investigate whether injected USSCs contribute to any component of regenerated skin, we applied immunohistochemical staining using antibodies against either human nuclei antigen or human-specific CD29, which are expressed in USSCs. However, the human cells were only sporadically identified in the dermis and the subcutis layer of the wound, but not in the epidermis or regenerated skin appendages (Fig. 6). Figure 6B and C demonstrate a representative detection of human cells in the dermis of a wound biopsy on day 8. Through immunohistochemical staining for endothelial protein CD31, we also concluded that USSC treatment promoted angiogenesis in the wound. Compared with the near absence of CD31 staining in the wound beds of the PBS-treated group, a number of CD31-positive cells (81 ± 23 per wound section, n = 5) were observed in the USSC-treated wounds at day 3 (data not shown). A significant difference in the number of CD31-positive cells was also observed between the two groups on day 8 (data not shown). These endothelial cells in the USSC-treated wound bed were of murine origin, since they were not stained positive for human-specific antigens (data not shown). This suggests that USSC treatment promoted angiogenesis in the wounds predominantly via a paracrine effect.

Keratin and human-specific antigen staining of wound site 40 days postinjection. (A) Immunoflurorescence staining of anti-KRT14 antibody (red) and anti-human-specific CD29 (INTβ1) antibody (green) in the wound section on day 40 post-USSC injection showed the KRT14-positive epidermis and skin appendages in the center of the wounds. There was no specific staining for human cells, except for the autofluorescence from the hair shafts. Scale bar: 50 μm. Immunostaining with human-specific nuclei antigen was also conducted, with similarly negative results. (B) Detection of human-specific CD29 (green) staining in the dermis of the wound 8 days postinjection. Skin sections were immunostained with anti-human-specific CD29 followed by staining with Alex 488-conjugated secondary antibody. Nuclei were stained by DAPI. (B) Merged image at a 10× magnification. Scale bar: 20 μm. (C) Higher magnification image on the area selected in (B). Scale bar: 20 μm.
In Vivo Bioluminescent Imaging (BLI) of Xenotransplanted USSCs
In the above wound healing model, USSCs were not directly injected in the excisional wound. To determine whether the sporadic staining of human cells in the wound was a result of specific migration or diffusion of the injected cells, we transduced USSCs with luciferase reporter gene for BLI (6). The modified USSCs exhibited the same set of cell surface markers as untransduced USSCs (data not shown). To confirm luciferase gene activity and specificity of BLI, the modified USSCs were serially diluted (from 1 million to 1,250 cells) and intradermally injected into mice, respectively. A linear correlation between the in vivo photon flux and cell dose was revealed by BLI, demonstrating the sensitivity and specificity of the imaging system (Fig. 7).

Bioluminescent imaging of injected USSCs. Green fluorescent protein/luciferase (GFP/Luc)-labeled USSCs, diluted from 1 million to 1,250 cells, were respectively injected intradermally into immunocompromised mice, to demonstrate a linear correlation between in vivo animal photon flux with the injection cell dose.
A total number of 1 million modified USSCs were then intradermally injected at multiple sites around the wound to analyze their temporal biodistribution relative to the wound (Fig. 8A–C). To our surprise, there was no distinguished migration of the cells to the wound bed, even when USSCs were injected at the edge (Fig. 8A, top left injection). The hot spot bioluminescence remained at the initial injection sites, despite the closure of the wound over time. However, a trail of the luminescent signal was observed, depicting the migratory track of USSCs from a more distant injection site toward the wound (Fig. 8B, depicted by an arrow). Such a trailing signal was not observed after day 13, when the wounds were almost closed. To enable visualization of low-level luminescence in the wound, the surrounding highly luminescent area was covered by XBP-24 black paper. Specific bioluminescent signal was then captured in the wound, most likely produced from the cells that were injected at the top left edge of the wound (Fig. 8D, bottom left panel). By shifting the blocking paper to image the right margin of the wound, a trailing bioluminescent signal was observed from the unwounded area to the wound (Fig. 8D, bottom right panel). This provided direct evidence for the specific migratory ability of locally injected USSCs to the wound. However, compared with the intense signal at the injection sites, only a minimal number of the cells trafficked to the wound. Temporal quantification on the total photon flux in the region (ROI, defined by a red rectangle) that included the wound and all the injection sites revealed a decrease of bioluminescence by 59.9% (±15.8%, n = 2) over 3 days and by 95.06% (±1.1%, n = 2) over 7 days, compared with the day 1 total photon flux (data not shown). The bioluminescent signal then gradually decreased to 1.07% (±0.42%, n = 2) in 1 month and maintained at 0.5–1% level until the end of the experiment (3 months).
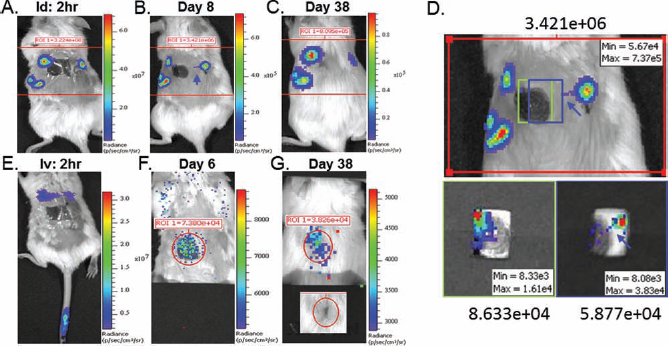
In vivo bioluminescent imaging of the mice with excisional wounds following injection. In vivo bioluminescent imaging of the mice with excisional wounds following GFP/Luc-labeled USSC intradermal (A–D) and tail vein intravenous (E–G) injection, with individual color scale. (A) Two-hour post-intradermal injection of 1 million USSCs. The total photon flux (p/s) at the region of interest (ROI) was 3.224e+08. (B) Day 8 post-USSC post-intradermal injection, with a total photon flux at ROI of 3.421e+06. The blue arrow depicts a migratory track of USSCs toward the wound. Further analysis of the ROI is illustrated in (D). (C) Day 38 post-USSC intradermal injection, with a total photon flux at ROI of 8.095e+05. (D) Top panel is a magnified image of ROI as shown in (B). The low luminescence from the wound, defined by the green (bottom left) and blue (bottom right) rectangle, was visualized after blocking the surrounding high-intensity luminescence. The blue arrow depicts a migratory track of the USSCs from a more distant injection site on the right toward the edge of the wound (E). Image taken at 2 h post-tail vein injection of 1 million USSCs. (F) D6 post-USSC intravenous injection. The bottom half of the mouse was blocked to exhibit a relative weak bioluminescent signal in the upper body. The total photon flux within the area of the wound was 7.390e+04. (G) Day 38 post-USSC intravenous injection. The total photon flux within the area of the wound was 3.826e+04.
We also explored the dynamics of USSCs following tail vein injection. Two hours postinjection, bioluminescent signals were observed in the lungs (illustrated by two distinct hot spots corresponding to the location of two lung lobes) and also in the tail, where the cells were injected (Fig. 8E). The signal from the lungs was detected on days 1, 2, and 3 postinjection and was absent after day 6, when a localized signal was captured in the wound bed (Fig. 8E–G). The bioluminescent signal from the wound represents real illumination from the injected cells, since such a localized signal was not present in the same parameter imaging using no-cell control mice with excisional wounding. It was also clear that the bioluminescence was generated from the cells in the skin, but not in any internal organs such as the liver or the spleen, as determined by the specific luminescence in the skin seen from the side view (data not shown). Based on the correlation between total photon flux and cell dose (Fig. 7), an estimate of 500–1,000 cells migrated to the skin wound on day 6. Similar biodistribution and migration of USSCs to the skin wound was also observed following intraorbital injection (data not shown). This demonstrated that USSCs have the ability to traffic to the site of injury through the circulation.
Discussion
Here we demonstrated the ability of CB-derived USSCs to promote skin regeneration. As USSCs and MSCs share overlapping features in immunophenotype and osteogenic/chondrogenic differentiation pathways, we analyzed the derived colonies for the expression of DLK-1, a marker to distinguish USSCs from MSCs (19). Consistent with the previous report (19), certain CB-derived colonies (three out of six) exhibited no expression of DLK-1 (Fig. 1A). Accordingly, a very few percentage of these cells were stained positive for AP activity (data not shown), compared with the DLK-1-expressing cells (Fig. 1C). Therefore, only the DLK-1-positive cells were termed USSCs and used in this study.
We demonstrated that USSCs share certain ES cell properties, including the expression of SSEA4 and AP in a subset of USSCs. However, USSCs lack the TRA1-60 surface marker and only weakly express Oct4 and Nanog. The incomplete DNA methylation on the regulatory regions of the Oct4 and Nanog genes may contribute to a basal level of gene transcription, and the treatment by DNA methylation inhibitor further activates their expression. Meanwhile, we also observed heterogeneity in the extent of DNA methylation among the bisulfitesequenced colonies; some individual cells were more unmethylated in the gene regulatory regions than others. It is unclear whether the heterogeneity, shown in the DNA methylation, expression of SSEA4, and AP activity, was a result of variation during single clonal expansion, impurities of staring colonies, or a combination of both. Recently, a method was described involving the use of cloning cylinders to derive single clonal populations from potentially multiple initiation colonies of the same CB unit (19). The clonal-derived cells appeared to be more defined and homogenous populations (19,28). However, their expressions of SSEA4 and AP activity were not reported. In our experimental settings, we trypsinized whole plates instead of individual initiation colonies. Therefore, our expanded cells may have started from more than one clone from each CB unit. We hypothesize that the USSCs homogenous for such stem cell signatures may have more efficient differentiation abilities and more significant beneficial effects in vivo.
In the full-thickness wounding model, we demonstrated that USSCs facilitated the rate of wound healing by promoting reepithelialization and angiogenesis. Improved skin histology with USSC-treated wounds, compared with the same time point PBS control, was observed at all the time points that were analyzed, as determined by the progression of epithelialization and formation of epidermis. One interesting observation is the presence of skin appendages in the regenerated wound of the USSC-treated mice. Although de novo formation of hair follicles has been reported (14), regeneration of hair follicles and other glands in wounds is rare and remains a challenge for wound therapy. It has previously been shown that transplantation of high-density MSCs (3 million) together with biomimetic nanofiber scaffold (NFS) in the wounding animal model resulted in the formation of hair follicles and sebaceous glands (26). In this current study, we demonstrated that intradermal injection of USSCs at a 1-cm distance from the edge of the wound also resulted in the formation of skin appendages at the center of the wounds. These structures were formed by endogenous keratinocytes, as demonstrated by their positive staining for KRT14 and lack of human-specific staining (Fig. 6). USSC-treated wounds also exhibited enhanced cellularity and increased vasculature at early days posttreatment. Angiogenesis is a critical component in the wound healing process. Formation of new blood vessels is essential to sustain the newly formed granulation tissue and the survival of keratinocytes. Under this experimental condition, USSCs were not found in the formed vascular structures within the wounds. This suggests that USSCs most likely promoted skin regeneration through a paracrine effect. USSCs have been demonstrated to constitutively produce a series of growth factors and cytokines, some of which, such as transforming growth factor 1β, granulocyte macrophage colony-stimulating factor, VEGF, and hepatocyte growth factor, have been used in studies for the treatment of diabetic wounds in animals and diabetic ulcers in patients (3,4,20,27,41. Moreover, these cytokines are produced at a significantly higher level in USSCs than MSCs (20). It is likely that USSCs released these growth factors to the wound, promoting epithelialization and regeneration of skin structures. Such a paracrine function of USSCs was recently reported in an acute animal model of spinal cord injury (30).
In the current study, we demonstrated the ability of USSCs to express hallmarks of keratinocyte differentiation genes under defined in vitro conditions. However, in vivo differentiation from the locally injected USSCs was not observed in the wounds. Several groups have previously reported transdifferentiation of BM-derived MSCs to multiple skin types, after either intravenous or direct injection/topical application to the wound (23,26,29,39). Some other groups, however, reported a lack of epidermal or endothelial differentiation from MSCs (2,31). The underlying reason for the disparities between the reports are unclear; however, at least two factors contribute to an efficient in vivo differentiation from any potential cell population, that is, accumulation of enough cells and an optimal microenvironment for differentiation. As suggested by both immunocytochemistry and BLI, the number of USSCs in the wound represented only a small percentage of the total injected cells. In addition, the loose connective tissue in the subcutis is likely the path for the migration of intradermally injected USSCs. The migrated cells thus may be restricted in their ability to incorporate into the epidermal layer, leading to an inefficient differentiation. Nevertheless, even with an absence of apparent in vivo skin differentiation, injection of USSCs exerted significant beneficial effects on wound healing and remodeling of epidermis. We are now investigating the conditions for the USSCs' in vivo differentiation, by a combination of USSC pretreatment in keratinocyte induction medium and direct topical administration on the wound.
The BLI results in this study revealed the dynamic information and specific migration of a minority of USSCs toward the wounding environment following both intradermal and systemic administration. It has been previously demonstrated that about 10% of the MSCs express chemokine receptor CCR7, and circulating MSCs were recruited to the wound site by the interaction between this receptor and chemokine SLC/CCL21 (29). Here we showed that about 10.76% of USSCs also express CCR7, which may be the driving force for the recruitment of USSCs to the site of injury. In addition, USSCs express PDGFRα. It has recently been demonstrated that lineage negative (Lin-)/PDGFRα+ cells from BM contribute to the regeneration of the epidermis after skin graft in vivo (32). High-mobility group box-1 has played a key role in mobilization and recruitment of Lin-/PDGFRα+ cells into regeneration of injured skin (32). Enriching USSCs for the cells with high expression of these chemokine receptors may enhance the migratory ability of USSCs. Meanwhile, the correlation between the expression of such chemokine receptors and the stem cell signatures SSEA-4 and AP will be determined. In future studies, topical application of chemoattractant proteins, CCL21, and high-mobility group box-1 on the wound may also facilitate the recruitment of USSCs, thus enhancing their beneficial role in skin regeneration.
In summary, we have characterized the genomic and functional properties of HUCB-derived USSCs and demonstrated their ability to promote wound healing. Our data also demonstrated the sensitivity of the BLI procedure to track the migration of a minority of the cells and identified strategies to further enhance the dermal regenerative potential of USSCs. Meanwhile, we demonstrated that USSCs express C7 protein that is defective in patients with RDEB. This study has raised the possibility of USSC's potential therapeutic value not only in wound healing but also in the treatment of patients with RDEB and has formed the basis for our future investigations of the HUCB-derived USSCs in a murine model of RDEB (Col7a1 knockout) (12).
Footnotes
Acknowledgments
We greatly acknowledge the technical assistance from Ms. Yan Lu at the SDRC at Columbia University and Dr. Aradhana Tiwari at NYMC. We thank Dr. Glenn Merlino at the National Cancer Institute for providing the pSico PolII-eGFP-Luc2 plasmid. We also thank Drs. David Bickers and David Owens at CUMC for the stimulating discussion and the support from coinvestigators at the Pediatric Cancer Research Laboratory at NYMC. We also acknowledge Erin Morris, RN, for assistance in the preparation of this article. This work was supported by NIH/NIAMS P30 AC44535 Skin Disease Research Center grant (SDRC), Debra International Foundation, and the Pediatric Cancer Research Foundation. The authors declare no conflict of interest.