
Abstract
The aim of this study was to identify novel angiogenic mechanisms underlying the regenerative process. To that end, interactions between adipose tissue-derived stromal cells (ASCs) and bone marrow cells (BMCs) were initially investigated using real-time fluorescence optical imaging. To monitor cell behavior in mice, we injected green fluorescent protein-positive (GFP+) BMCs into the tail vein and injected PKH26-labeled ASCs behind the ears. Angiogenesis and inflammation were observed at these sites via an optical imaging probe. Injected GFP+ BMCs migrated from the blood vessels into the tissues surrounding the ASC injection sites. Many of the migrating GFP+ BMCs discovered at the ASC injection sites were inflammatory cells, including Gr-1+, CD11b+, and F4/80+ cells. ASCs cocultured with inflammatory cells secreted increased levels of chemokines such as macrophage inflammatory protein (MIP)-1α, MIP-1β, keratinocyte-derived chemokines, and monocyte chemotactic protein 1. Similarly, these ASCs secreted increased levels of angiogenic growth factors such as hepatocyte growth factor and vascular endothelial growth factor. However, when anti-CXC chemokine receptor type 4 antibody was injected at regular intervals, the migration of GFP+ BMCs (especially Gr-1+ and CD11b+ cells) to ASC injection sites was inhibited, as was angiogenesis. The collective influence of the injected ASCs and BMC-derived inflammatory cells promoted acute inflammation and angiogenesis. Together, the results suggest that the outcome of cell-based angiogenic therapy is influenced not only by the injected cells but also by the effect of intrinsic inflammatory cells.
Introduction
As a treatment strategy in the field of regenerative medicine, cell-based therapy has been shown to be useful for treating vascular disorders such as ischemic injury (2,40). There are several reasons for its beneficial effects, including the ability of transplanted cells to differentiate into blood vessels (33,45) and secrete angiogenic growth factors (16). In addition, endothelial progenitor cells (EPCs) that circulate in the blood can respond to signals from damaged tissues (39). However, in many circumstances, these responses alone may not account for the observed effects (29). Despite significant advances in our understanding of the mechanisms involved in angiogenesis, numerous steps remain to be identified and elucidated.
Adipose tissue is a useful source for cell-based therapy. It is composed of several heterogeneous cell types including vascular, endothelial, and stem cells, which has paracrine effects from angiogenic growth factors such as hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), and fibroblast growth factor-2 (FGF-2) (24,30,36). Intravenous injections of adipose tissue- derived stromal cells (ASCs) have been shown to improve the recovery of hindlimb function in mice and rats following ischemic vascular injury (30,33). However, multiple different cell types are used for angiogenic therapy, and the prognosis following cell transplantation has proven to be inconsistent.
The process of wound healing is divided into three phases, namely, inflammation, proliferation, and remodeling. Each phase overlaps and is influenced by several factors, including cytokines, growth factors, and chemokines (43). The inflammation phase involves a rapid and highly controlled interplay of various cells and soluble mediators of inflammation, coagulation, immunity, and angiogenesis. Cells related to the inflammation phase include platelets, neutrophils, monocytes, and macrophages. These cells leave the circulation and migrate to the site of injury. Stem cells from the bone marrow or elsewhere also migrate to the site of injury where they differentiate into required cells (7,27,32). Alternatively, they do not differentiate into required cells but rather remain as stem cells and secrete trophic factors (22). Chemokine (C-X-C motif) receptor type 4 (CXCR4) reportedly plays a major role in the mechanism of wound healing (32), in which it appears to be capable of mobilizing hematopoietic stem cells into the blood as peripheral blood stem cells (26). The ligand for CXCR4 is stromal-derived factor-1 (SDF-1) (11). The combination of CXCR4 and SDF-1 mediates the ischemia-induced recruitment of circulating bone marrow-derived stem/progenitor cells to the site of injury. These mobilized cells contribute directly to vessel growth (3,47). Bone marrow-derived EPCs circulating in the blood also reach the site of injury because of SDF-1 and are responsible for stabilizing nascent blood vessels (42).
Chemokines were originally identified for their roles in leukocyte recruitment. They are now known to also facilitate neovascularization in patients with various diseases, including cancer, fibroproliferative disorders, and ischemic heart disease (10,14). In addition, the expression of chemokine receptors on resident cells suggests that chemokines contribute to the regulation of tissue remodeling and angiogenesis (23). These effects appear to be secondary to recruitment of inflammatory cells, including neutrophils, monocytes, and macrophages. We speculated that angiogenesis induced by cell therapy may be attributable to factors other than those identified previously.
In the real-time fluorescence optical imaging-based study presented here, we sought to identify novel angiogenic mechanisms, particularly those occurring during the acute phase response of ASC-based therapy.
Materials and Methods
Preparation of Mouse ASCs
This study was conducted in accordance with the guidelines for the care of animals as defined by the Institutional Review Board of the National Defense Medical College (Saitama, Japan).
Primary ASCs were prepared from the inguinal adipose tissue of 8-week-old male C57BL/6 mice (n = 46; Japan SLC, Shizuoka, Japan). Adipose tissue was washed extensively with Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) and digested for 120 min at 37°C with 0.1% collagenase type I (Wako Pure Chemical Industries, Ltd., Osaka, Japan). Samples were then resuspended in DMEM, passed through a 40-μm nylon mesh (BD Falcon, Bedford, MA, USA), and centrifuged at 350 × g for 10 min. Cell pellets were suspended for 5 min in lysis buffer (Sigma-Aldrich) and centrifuged at 350 × g for 10 min. Cells were then suspended in DMEM containing 5% heat-inactivated fetal bovine serum (FBS; Cambrex Co., East Rutherford, NJ, USA) and antibiotics (Sigma-Aldrich; control medium). These cells were used as ASCs. For each experiment, we used a cell population that was passaged twice. Cell numbers were determined using a hemocytometer (ERMA Inc., Tokyo, Japan) after trypsinization (Sigma-Aldrich).
Flow Cytometric Analyses
At least 2 × 105 cells were suspended in 30 μl phosphate-buffered saline (PBS; Sigma-Aldrich) containing 2% FBS and incubated for 20 min with fluorescence-labeled antibodies specific for cluster of differentiation 29-phycoerythrin (CD29-PE), stem cell antigen 1 (Sca-1)-PE, CD117-PE, CD34-PE, CD11b-PE, CD45-PE, CD133-PE, granulocyte receptor-1 (Gr-1)-PE, F4/80-PE, CXCR4-PE, CD73-PE, CD90.2-PE, CD105-PE, and CD31-fluorescein isothiocyanate (FITC) or CD106-FITC (Beckman Coulter, Fullerton, CA, USA). Nonspecific fluorescence was determined by control immunoglobulin G for each isotype (all antibodies except CD106; eBioscience, San Diego, CA, USA).
Isolation of Inflammatory Cells and Bone Marrow Cells (BMCs)
Intraperitoneal injections of male C57BL/6-Tg [CAG-enhanced green fluorescent protein (eGFP)] mice (Japan SLC) (8–13 weeks) (n = 20) were performed with 2 ml of 12% sodium casein (Wako Pure Chemical Industries, Ltd.) in saline. Gr-1+ cells recruited to the intraperitoneal space were collected by laparotomy 1 day after induction, and recruited CD11b+ cells were collected 7 days after induction. BMCs were collected by flushing the femurs and tibias. Cell populations were separated with high purity (>97%) using a magnetic cell-sorting method (Miltenyi Biotec, Bergisch Gladbach, Germany) with antibodies against CXCR4, Gr-1, and CD11b.
Migration Assay
BMC migration was evaluated at 24 h using a 96-multiwell insert system (BD Biosciences, San Jose, CA, USA) and a fluorescence microplate reader (Fluostar Optima; BMG Labtech, Tokyo, Japan). BMCs (2.0 × 104 cells/50 μl) were loaded onto the upper compartment of a transwell chamber, whereas the lower compartment contained ASC culture supernatant. ASCs were plated at 5.0 × 104 cells/well with 200 μl of control medium in 48-well culture plates, and the supernatant was collected after 2 days. To measure the inhibitory effect of neutralizing antibody on BMC migration, 20 ng of anti-mouse CXCR4 antibody (R&D Systems, Minneapolis, MN, USA) was added to the upper compartment with the BMCs. DMEM containing 5% FBS was used as a control.
Detection of Growth Factors, Cytokines, and Chemokines
Cells were plated at 5.0 × 104 cells/well in 48-well culture plates (Sumilon, Tokyo, Japan). When cocultured with inflammatory cells, ASCs were plated and allowed to attach. The next day, CXCR4+, Gr-1+, and CD11b+ cells were plated at the same cell number. After 2 days of culture, the concentrations of secreted proteins in the supernatants were measured using Bio-Plex [Bio-Rad, Hercules, CA, USA; macrophage inflammatory protein (MIP)-1α, MIP-1β, MIP-2, keratinocyte-derived chemokine (KC), VEGF, and FGF-2] and enzyme-linked immunosorbent assays [SDF-1 (R&D Systems), HGF (Institute of Immunology, Tokyo, Japan), and monocyte chemotactic protein (MCP)-1 (Thermo Fisher, Boston, MA, USA)].
In Vivo Imaging
After careful removal of hair from the ears of the recipient mice using a depilatory cream (Kracie Holdings, Tokyo, Japan), the animals were irradiated with 6 Gy using X-ray irradiation equipment (MBR-1505R2; Hitachi Medical Co., Tokyo, Japan). Care was taken to avoid damage to the ears. BMCs were collected by flushing the femurs and tibias of male C57BL/6-Tg (CAG-eGFP) mice (8–13 weeks; n = 45). These cells (2.0 × 107 cells/mouse) were injected into the tail veins of recipient mice 24 h after irradiation. Movement of GFP+ cells introduced into the circulation was observed using a fluorescence optical imaging system (OV110; Olympus, Tokyo, Japan) (18). Anesthesia was induced by inhalation of 1.5% isoflurane (Abbott Japan Co. Ltd., Tokyo, Japan) in oxygen. The next day, ASCs or PKH26-labeled ASCs (Sigma-Aldrich; labeled according to manufacturer's instructions) were in PBS, and 5 μl (2.0 × 106 cells/ear) was injected behind the ears of the mice using a 30-gauge insulin syringe (Becton Dickinson, San Jose, CA, USA). Elizabethan collars (Natsume Seisakusho, Tokyo, Japan) were placed on the necks of the mice to avoid ear scratching. Images of angiogenesis were captured within 15 min following the tail vein injection of 100 μl of an optical contrast agent (AngioSense-IVM 750; VisEn Medical, Bedford, MA, USA). Quantification of injected PKH26-labeled cells and migrated GFP+ cells was accomplished by using the OV110 system. Anti-mouse CXCR4 antibody (100 μg/100 μl; R&D Systems) was injected into the tail vein 1 h before the cells were injected into the ear. The same amount of antibody was injected on the following day and on every other day thereafter.
Immunostaining
For immunostaining of endothelial cell markers in vitro, such as CD31 as a measure of tube formation, ASCs were cultured in 5% FBS and DMEM on 48-well plates for 7 days. Cells fixed with 4% paraformaldehyde (PFA; Wako Pure Chemical Industries, Ltd.) were incubated for 1 h with antibodies against CD31 (1:100 dilution; BioLegend, San Diego, CA, USA) and for 30 min with anti-rat IgG antibodies (R&D Systems). This was followed by staining with diaminobenzidine (Dako A/S, Glostrup, Denmark). For tissue sampling, mice were anesthetized with nembutal (Dainippon Sumitomo Pharma Co., Ltd, Osaka, Japan) and perfusion fixed with 4% PFA. Their ears were cryoembedded in optimum cutting temperature compound (Sakura Finetek, Tokyo, Japan) and sectioned (10 μm thickness). Cryosections were refixed with 4% PFA for 15 min and incubated for 1 h at room temperature with 10% FBS in PBS. After blocking, cryosections were incubated for 1 h with primary biotinylated antibodies specific for Gr-1, CD11b, F4/80, CD31, and vascular endothelial (VE)-cadherin (all at 1:50 dilution; eBioscience). The cryosections were then incubated for 30 min with allophycocyaninconjugated streptavidin (1:50 dilution; eBioscience). Hoechst 33258 (Dojindo Laboratories, Kumamoto, Japan) was used for nuclear staining.
Statistical Analyses
Data are expressed as means±standard deviation (SD). Two-group comparisons were analyzed by the unpaired two-tailed t test. Multiple comparisons were evaluated by ANOVA, Tukey's test, or Dunnett's test as appropriate. A value of p < 0.05 was considered statistically significant.
Results
Characterization of ASC Populations
To characterize the twice-passaged ASC populations, we analyzed cellular antigens by flow cytometry and used primary ASCs for comparison (Table 1). Freshly divided primary ASC cultures were heterogeneous, containing hematopoietic cells, adherent cells, and endothelial cells. When the cells were subcultured twice, many of them expressed typical surface antigens, including CD29, Sca-1, CD90.2, CD105, and CD73. The cells were negative for markers such as CD34, CD117, Gr-1, CD11b, CD31, CD106, CD133, CD45, F4/80, and CXCR4. Primary ASCs formed endothelial tubes, whereas twice-passaged ASCs did not (Fig. 1). All CD29-containing integrins bind to extracellular matrix proteins, including collagen, laminin, fibronectin, and vitronectin, whereas some CD29-containing integrins also interact with cell receptors (31). Sca-1 is a member of the lymphocyte antigen 6 multigene family of type V glycophosphatidylinositol-anchored cell surface proteins. CD73, CD90, and CD105 are expressed on mesenchymal cells (1,12). Based on the primary functions of the cell surface markers identified, these results suggest that the twice-passaged ASCs included a large population of adherent cells such as mesenchymal cells, but not hematopoietic cells or mature endothelial cells.
Expression of Cell Surface Markers in Twice-Passaged (P2) and Primary (P0) Adipose Tissue-Derived Stromal Cells (ASCs)

Percentages of positive cells for each surface marker are shown. CD29, cluster of differentiation 29; Sca-1, stem cell antigen 1; Gr-1, granulocyte receptor 1; CXCR4, chemokine (C-X-C motif) receptor 4. Values are mean±SD.
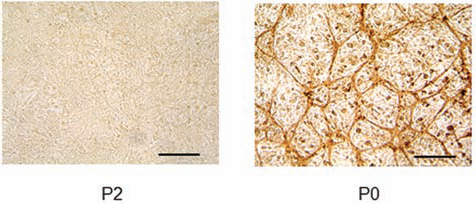
Tube formation capability of twice-passaged adipose tissue-derived stromal cells (ASCs; P2) and primary ASCs (P0). Cultured cells for 7 days were immunostained with anti-cluster of differentiation 31 (CD31) antibody, a marker for endothelial cells. Scale bar: 100 μm.
Cell Behavior of Injected Cells by In Vivo Real-Time Fluorescence Optical Imaging
To demonstrate the interactions between injected ASCs and BMCs in vivo, we followed the procedure outlined in Figure 2A. Almost all ASCs were labeled with PKH26 (Fig. 2B). After injecting GFP+ BMCs into the tail veins of irradiated mice, we captured images of these cells circulating in the blood vessels using a real-time fluorescence optical imaging system (Supplemental Movie I; http://www.geocities.jp/ct_0768_supple).
GFP+ BMCs began migrating toward the injection sites approximately 6 h after PKH26-labeled ASCs were injected into the ear. The number of GFP+ BMCs migrating toward these sites increased gradually with time (Fig. 2C, top). GFP+ BMCs circulating in the blood vessels readily adhered to the endothelium of tissues surrounding the injected ASCs, and the adhered BMCs moved via extra-vascular migration (Fig. 2D, left; Supplemental Movies II and III; http://www.geocities.jp/ct_0768_supple). Sequential observations showed that many GFP+ BMCs had transmigrated through the vessel walls into the surrounding tissues over the course of 1 week (Fig. 2D, right; Supplemental Movie IV; http://www.geocities.jp/ct_0768_supple). When both ASCs and anti-CXCR4 antibody were injected, most GFP+ BMCs did not migrate to the ASC injection sites for 3 days. However, GFP+ BMCs began to migrate approximately 5 days after injection, and they were present at the injection sites 7 days after injection (Fig. 2C, middle). Only a few GFP+ BMCs migrated toward the injection site if saline was used as a control (Fig. 2C, bottom). Although ASCs were injected, BMC migration was strongly inhibited by blocking CXCR4 for 3 days. However, 5 days after ASC transplantation, BMC migration was no longer controlled by antibody-mediated blocking of CXCR4 (Fig. 2E).
To confirm the presence of injected ASCs, the cells were labeled with PKH26, and their fluorescence intensity was measured. When ASCs were injected alone, the PKH26-positive area reached a maximum at 3 days and decreased thereafter. When anti-CXCR4 antibody was injected, the PKH26-positive area did not decrease, even after 7 days (Fig. 2F and G). PKH26 fluorescence intensity was observed at the injection sites for more than 1 month, with or without anti-CXCR4 antibody injection (data not shown).
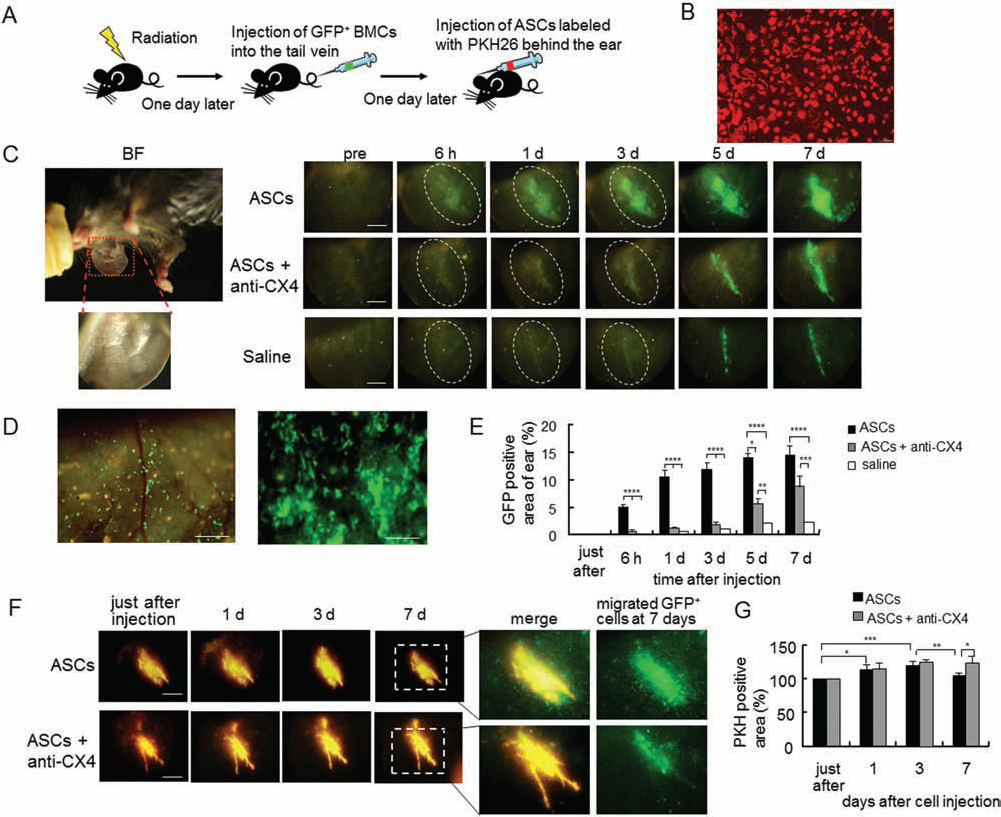
Observation of cell behavior after cell injection using in vivo real-time fluorescence optical imaging. (A) Procedure for realtime fluorescence optical imaging utilized for observing the interactions between injected PKH26-labeled ASCs and green fluorescent protein-labeled bone marrow cells (GFP BMCs). (B) ASCs labeled with PKH26. (C) Time-dependent patterns of GFP+ BMCs migrating to the site of ASC injection. Top: Images of ASCs injected into the ear. GFP+ cells began to migrate gradually toward the injection site after approximately 6 h. Peak brightness was observed at 7 days. Middle: Images of the injected ASCs and anti-chemokine (C-X-C motif) receptor 4 (CXCR4; CX4) antibody. Bottom: Images of saline injection. ASCs or saline was injected behind the ear. The injection site is shown by a dotted circle. pre, before injection; BF, bright field; others are fluorescence images. Scale bar: 2 mm. (D) Left: Transmigration through the vessel wall of GFP+ BMCs 6 h after ASC injection. Scale bar: 500 μm. Right: Transmigration of many GFP+ BMCs through the vessel wall into the surrounding tissues 7 days after injection. Scale bar: 200 μm. (E) Quantification of migrated GFP+ cells at the sites of injection of ASCs or saline (n = 5). *p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001; Tukey's test. (F) Time-dependent appearance of PKH26-labeled ASCs injected into the ear. Top: Images of the injected ASCs. Bottom: Images of the ASCs injected after anti-CXCR4 antibody. The second right panels show merged images of PKH26-labeled ASCs and migrated GFP+ BMCs at 7 days. Scale bar: 2 mm. (G) Quantification of injected ASCs. Fluorescence intensity was calculated immediately after injecting PKH26-labeled ASCs (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001; Tukey's test.
Relationship Between CXCR4+ Cells in BMCs and ASCs In Vitro
CXCR4 expression in BMCs was approximately 36% in vitro (Fig. 3A). BMCs showed a migrational response to ASC supernatant, and migration was inhibited by antibody-mediated blocking of CXCR4 (Fig. 3B). ASCs had the ability to secrete SDF-1, whereas CXCR4+ cells did not (Fig. 3C). ASCs secreted many chemokines; moreover, coculturing with CXCR4+ cells led to high levels of chemokine secretion, including MIP-1α, MIP-1β, KC, and MCP-1 (Fig. 3D).

Relationship between CXCR4+ cells in BMCs and ASCs. (A) Proportion of CXCR4+ cells in BMCs (36±4%) as determined by flow cytometry. (B) Migration activity of GFP+ BMCs in response to supernatants of ASC culture and subsequent inhibition by blocking of CXCR4 (CX4). Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal bovine serum (FBS) was used as a control. *p < 0.05, **p < 0.01; Tukey's test. (C) Amount of stromal-derived factor-1 (SDF-1) in supernatants of ASC culture (ASCs), CXCR4+ cell culture (anti-CX4), and coculture of ASCs with CXCR4+ cells (ASC + anti-CX4). N.D., not detected. (D) Amount of chemokines under each condition: ASC culture, CXCR4+ cell culture, and coculture of ASCs with CXCR4+ cells. MIP, macrophage inflammatory protein; KC, keratinocyte-derived chemokine; MCP-1, monocyte chemotactic protein-1. *p < 0.05, **p < 0.005 versus only ASC culture; Dunnett's test (n = 5).
Many Migrating BMCs Are Inflammatory Cells
CXCR4 signaling profoundly modulates angiogenesis and homing capacity of EPCs (42). Most injected BMCs within the extravascular site did not form an obvious blood vessel 7 days after ASC injection (Fig. 2D). CXCR4 is expressed in several cell types, including neutrophils, monocytes, EPCs, and hematopoietic precursor cells (13,41). ASCs have a strong potential for causing chemotactic effects by attracting inflammatory cells (Fig. 3D). Gr-1+, CD11b+, and F4/80+ cells represent granulocytes or neutrophils (6), monocytes (4), and macrophage (19), respectively. The rates of CXCR4+/Gr-1+ and CXCR4+/CD11b+ expression on BMCs were approximately 22.7% and 19.7%, respectively (Fig. 4A). Therefore, we used immunostaining to determine if the migrated GFP+ BMCs included inflammatory cells. One day after ASC injection, many migrated GFP+ cells were positive for Gr-1 and CD11b; however, there were very few Gr-1+ cells at 7 days (Fig. 4B). Instead, there were many F4/80+ cells at 7 days (Fig. 4C), which were not present 1 day after ASC injection (data not shown). The results suggest that the earliest migrated cells were granulocytes, neutrophils, and monocytes, followed by macrophages.
When anti-CXCR4 antibody was injected, few GFP+ BMCs or inflammatory cells migrated to the injection site at 1 day after injection (Fig. 4B). Blocking of CXCR4 inhibited the migration of neutrophils, granulocytes, and monocytes derived from the injected BMCs. Injection of ASCs induced an exaggerated acute phase inflammatory response, whereas blocking of CXCR4 inhibited this acute response. ASC supernatant induced the high migration of Gr-1+ and CD11b+ cells (Fig. 4D).

Characterization of GFP+ BMCs in the areas surrounding the injected ASCs. (A) CXCR4+/granulocyte receptor 1-positive (Gr1+) and CXCR4+/cluster of differentiation 11b-positive (CD11b+) cells in BMCs as detected by flow cytometry. The numbers in the graphs signify the expression of markers. (B) Cryosections 1 day after injecting ASCs, immunostaining of Gr-1 (red) and CD11b (red), migrated GFP+ BMC cells (green), and nuclei-stained with Hoechst 33258 (blue). Scale bars: 100 μm. Right graphs show the number of each cell marker (Gr-1 and CD11b)-positive cells, double-positive cells, and both cell marker (Gr-1 and CD11b)- and GFP-positive cells in the surrounding areas injected with ASCs at 1 and 7 days. For each section, cell numbers in three fields were counted, and mean values were calculated. N.D., not detected. *p < 0.05, **p < 0.01; t test. (C) Cryosections 7 days after injecting ASCs, immunostaining of F4/80 (red). Scale bars: 50 μm. (D) The migration activity of Gr-1+ or CD11b+ cells in the ASC supernatant. DMEM containing 5% FBS was used as a control. *p < 0.001, t test (n = 5).
Secretion of Chemokines by ASCs Is Enhanced by Interactions with Migrated Inflammatory Cells
To investigate the interactions between inflammatory cells such as Gr-1+ or CD11b+ and ASCs, we cocultured ASCs with inflammatory cells and measured the amounts of chemokines in the culture supernatants. ASCs have a high capability of secreting chemokines on their own. By coculturing with inflammatory cells, increased secretion of chemokines from several to tens of thousands of times was observed (Fig. 5A). Supernatants from cocultures of ASCs with inflammatory cells increased the migration activity of Gr-1+ or CD11b+ cells more than the supernatants from cultures of ASCs alone (Fig. 5B). Thus, the interaction of ASCs and inflammatory cells induced ASCs to secrete a large amount of chemokines. These secreted chemokines subsequently led to the migration of excessive numbers of inflammatory cells.

Secretion of chemokines by interactions between ASCs and inflammatory cells. (A) Quantity of chemokines under each condition, ASCs cocultured with Gr-1+ or CD11b+ cells, and each cell type alone. A, ASCs; G, Gr-1+ cells; C, CD11b+ cells. *p < 0.05, **p < 0.005 versus ASC single culture; Dunnett's test (n = 5). (B) Migration activity of Gr-1+ or CD11b+ cells in the supernatant of the ASC coculture with Gr-1+ or CD11b+ cells. *p < 0.001 versus ASC single culture; Dunnett's test (n = 5).
Angiogenesis Induced by Interactions Between ASCs and Inflammatory Cells
To examine angiogenesis after ASC injection, we employed an in vivo imaging strategy using an optical contrast agent (Fig. 6A). On day 7, images of primarily inflammation rather than angiogenesis were captured at the ASC injection area. However, the inflammation and angiogenesis areas were decreased by the presence of anti-CXCR4 antibody. Images were not captured at the saline injection area (control). Many GFP+ inflammatory cells accumulated at the site of angiogenesis and inflammation (Fig. 6A, second right panel). The level of angiogenesis and inflammation was increased at the site where GFP+ cells were accumulated rather than at the site where ASCs were injected (Fig. 6A, right). Angiogenesis and inflammation, as well as the migration of GFP+ cells, were inhibited by antibody-mediated blocking of CXCR4.
Positive identification of angiogenesis was accomplished via immunostaining of the endothelial cell markers CD31 and VE-cadherin (Fig. 6B and C). CD31+ and VE-cadherin+ cells were present in the tissue surrounding the location of GFP-positive cells in the ASC injection area but were absent from the central tissue of the ASC injection area at 7 days (Fig. 6D). The percentage of GFP+/CD31+ cells and GFP+/VE-cadherin+ cells around ASC injection sites was less than 5.0% and less than 0.2%, respectively, at 7 days. Cells that were double positive for PKH26 and CD31 or VE-cadherin did not exist. ASCs did not form vessels, and BMC-derived vessel-like structures were few. Angiogenesis was induced within the tissue surrounding GFP-positive cells at the ASC injection area after 7 days, but no blood vessels formed within the central portion of the injected ASC clusters, which were composed primarily of inflammatory cells. Furthermore, the number of vessels around the ASC injection sites decreased significantly with injection of anti-CXCR4 antibody (Fig. 6B and C). To investigate the relationship between ASC injection and the processes of angiogenesis and inflammatory cell migration, we measured the amount of angiogenic growth factors such as VEGF, HGF, and FGF-2 in supernatants obtained by coculture. While ASCs had a natural ability to secrete angiogenic growth factors, coculture with inflammatory cells was sufficient to increase VEGF and HGF secretion even further (Fig. 6E).
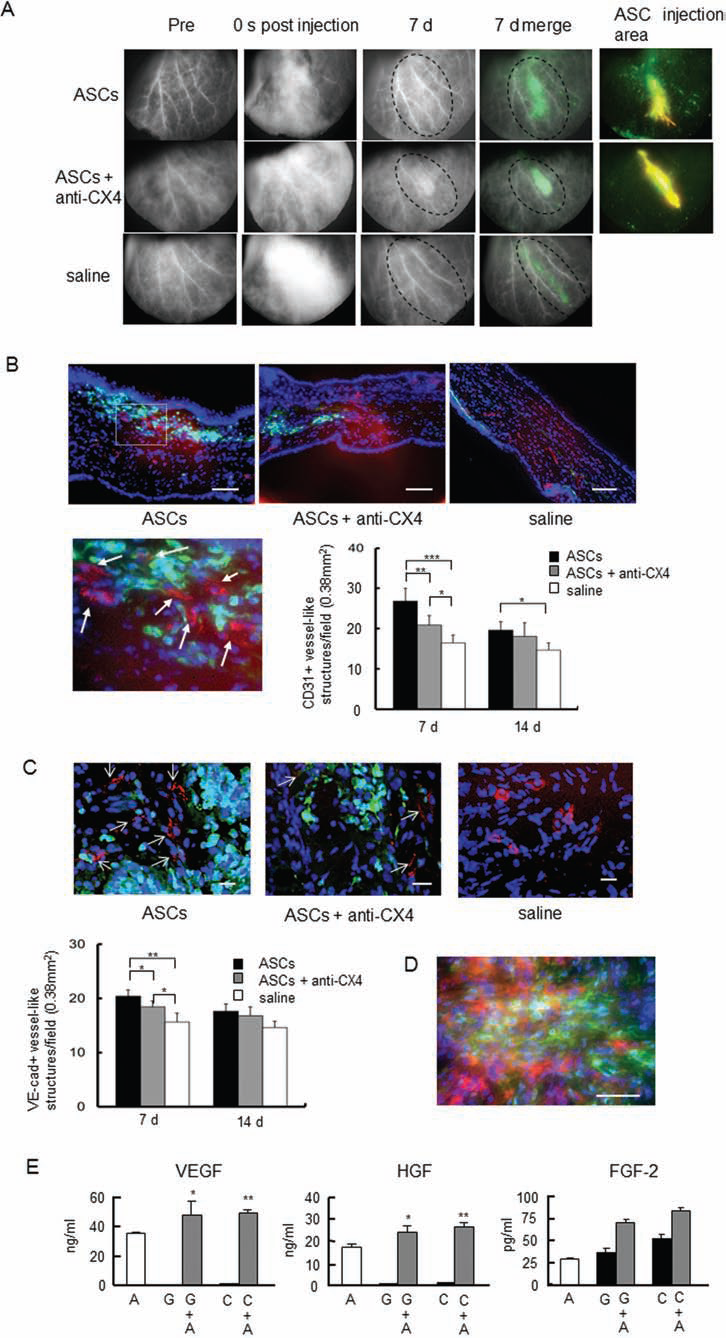
Angiogenesis induced by interactions between ASCs and inflammatory cells. (A) Changes in angiogenic and inflammatory patterns with time after ASC injection. Top: Changes over 7 days after ASC injection. Middle: ASCs and anti-CXCR4 (CX4) antibody injection. Bottom: Saline injection as control. An optical imaging probe was used as the angiographic agent. The second right panels show merged images of angiogenesis and inflammation and migrated GFP cells on day 7. The right panels show the ASC injection area. Scale bar: 2 mm. (B) Immunostaining of CD31+ cells (red; arrows) in the surrounding areas of injected ASCs 7 days after injection of ASCs or saline, migrated GFP+ cells (green), and nuclei stained with Hoechst 33258 (blue). Scale bar: 100 μm. The graph shows the number of CD31+ vessel-like structures per field (0.38 mm2). For each section, vessel-like structure numbers in five fields were counted, and mean values were calculated. *p < 0.05, **p < 0.01, ***p < 0.001; Tukey's test. (C) Immunostaining of vascular endothelial (VE)-cadherin+ cells (red; arrows) in the surrounding areas of injected ASCs 7 days after injection of ASCs or saline, migrated GFP+ cells (green), and nuclei stained with Hoechst 33258 (blue). Scale bar: 20 μm. The right graph shows the number of VE-cadherin (VE-cad)+ vessel-like structures per field (0.38 mm2). For each section, vessel-like structure numbers in five fields were counted, and mean values were calculated. *p < 0.05, **p < 0.001; Tukey's test. (D) Morphology of PKH26+ ASCs (red) and GFP+ cells (green) in the central part of the ASC clusters at 7 days. Scale bar: 50 μm. (E) Secretion of angiogenic growth factors under each condition, ASCs cocultured with Gr-1+ or CD11b+ cells, and each cell type alone. A, ASCs; G, Gr-1+ cells; C, CD11b+ cells; VEGF, vascular endothelial growth factor; HGF, hepatocyte growth factor; FGF-2, fibroblast growth factor-2. *p < 0.05, **p < 0.01 versus ASCs; Dunnett's test (n = 5).
Discussion
Inflammation is an important part of the wound healing process because it involves the induction of inflammatory factors and accumulation of various types of inflammatory cells (17). Inflammatory factors and cells facilitate tissue regeneration by replenishing a variety of cell types and extracellular components. We hypothesized that the tissue-regenerative properties of cells would be stimulated not only by the injected cells for the purpose of cell therapy but also by the BMCs and inflammatory cells that received signals from the injected cells and migrated toward them.
According to our histological results, many GFP+ inflammatory cells, including granulocytes, neutrophils, monocytes, or macrophages, were present at ASC injection sites. Interactions among these cells initiated an inflammatory reaction, which in turn induced the migration of many more inflammatory cells derived from the bone marrow. Moreover, the interactions of the injected ASC cells with the migrating inflammatory cells induced the secretion of additional chemokines. The release of these chemokines led to increased recruitment of inflammatory cells and the initiation of an extensive wound-healing process, which included angiogenesis. Recent studies have provided evidence that confirmed the role of chemokines in the recruitment of inflammatory cells to wound sites (15,34). MIP-1α and MIP-1β activate neutrophils and monocytes/macrophages, resulting in acute neutrophilic inflammation (28,44,46). MIP-2 and KC are expressed at sites of tissue inflammation after injury and/or infection (21) and are capable of activating and recruiting neutrophils (28). MCP-1 recruits monocytes and other cell types to sites of tissue injury and/or infection (10,28,46). Cell migration is a basic phenomenon exhibited by a wide variety of cell types involved in angiogenesis, inflammation, wound healing, and many other complex physiological processes. It is particularly important for the purpose of increasing the number and enhancing the function of neutrophils at wound sites during the inflammatory stage of wound healing. Our results demonstrated the importance of such inflammatory events during the process of angiogenesis. Injected cells are continually exposed to inflammatory cells, starting with neutrophils, granulocytes, and monocytes. Therefore, the injected cells continuously secrete many chemokines and angiogenic growth factors. Regarding a possible mechanism relevant to cell therapy, injected cells may have a pump-priming effect on wound healing via an inflammatory response.
Chemokines such as KC, MIP-2, and MCP-1 not only work as chemotactic factors for inflammatory cells but also work as angiogenesis factors (14). MCP-1 has been associated with the induction of VEGF-A gene expression. The induced VEGF then induces angiogenesis (20). The angiogenic activity of KC and MIP-2 appears to be neutrophil dependent because neutrophil depletion completely abrogates the angiogenic response (5). MIP-2 induces neutrophil-dependent angiogenesis by activation of VEGF-A (35). ASCs activated the secretion of KC, MIP-2, and MCP-1 by interaction with inflammatory cells (Fig. 5). Angiogenesis is influenced by angiogenic factors induced by chemokines, meaning that angiogenesis resulting from ASC injection will also likely be subject to the influence of angiogenic factors induced by chemokines. The injected ASCs did not form vessels, while the migrated BMC-derived cells formed a few veins (Fig. 6). Moreover, new vessel-like structures existed in the tissue surrounding the ASC injection site but did not exist in the central region of the injected ASC cluster. Therefore, ASC injection appears to induce angiogenesis from existing vessels in the surrounding tissue.
However, even if there are cells having a great pump-priming effect, the inflammatory response cannot start without receiving the appropriate signals. SDF-1 plays a pivotal role in angiogenesis (25) and mediates the homing of stem cells from the bone marrow by associating with circulating CXCR4+ cells (8,9). CXCR4 is highly expressed by neutrophils, monocytes, and early hematopoietic progenitor cells in the bone marrow (13,41). Wound healing is inhibited if SDF-1 or CXCR4+ cells are blocked in ischemic tissues (8). In a study, bone marrow-derived mono-nuclear cells expressing CXCR4 increased tissue invasion and restored blood flow in mice with hind limb ischemia (37). Our results showed that the migration of BMCs, including Gr-1+ and CD11b+ cells, was inhibited 3 days after CXCR4 was blocked, even if ASCs were injected. This resulted in a delayed inflammatory process, which subsequently resulted in the inhibition of angiogenesis. Blocking of CXCR4 controlled the migration of BMCs, including neutrophils and monocytes, during the short period of approximately 3 days. Therefore, the blocking of CXCR4 inhibited the acute inflammatory phase. Despite the presence of secreted chemokines from ASCs, CXCR4+ BMCs could not receive the necessary signals because of CXCR4 blocking. Therefore, it was confirmed that the function of CXCR4+ BMCs was important for the initiation of the inflammatory response during cell-based angiogenic therapy, as well as during wound healing. Sheikh et al. suggested that successful therapy in the ischemic myocardium may be dependent on modulation of the host environment and the transcriptional responses of transplanted cells (38). We speculate that the results of cell injection therapy may be dependent primarily on the recipient's immune response rather than the angiogenic capabilities of injected cells.
In conclusion, we demonstrated that cell-based therapy induced angiogenesis, which was dependent on both angiogenic growth factors and chemokines stemming from the interactions of injected cells with bone marrow-derived inflammatory cells. Furthermore, if an inflammatory response involving the migration of CXCR4+ cells is blocked, it can reduce angiogenesis even if cell-based therapy is administered.
Footnotes
Acknowledgments
This research was partially supported by the Ministry of Education, Culture, Sports, Science, and Technology, Japan, Grant-in-Aid for Young Scientists (B), 22700486. We thank Ms. K. Higuchi (Olympus) for her technical suggestions and Ms. N. Ishida for proof checking the manuscript. The authors declare no conflicts of interest.