
Abstract
Leukodystrophies are a group of disorders characterized by myelin dysfunction, either at the level of myelin formation or maintenance, that affect the central nervous system (CNS) and also in some cases, to a lesser extent, the peripheral nervous system (PNS). Although these genetic-based disorders are generally rare, all together they have a significant impact in the society, with an estimated overall incidence of 1 in 7,663 live births. Currently, there is no cure for leukodystrophies, and the development of effective treatments remains challenging. Not only leukodystrophies generally progress very fast, but also most are multifocal needing the simultaneous targeting at multiple sites. Moreover, as the CNS is affected, the blood–brain barrier (BBB) limits the efficacy of treatment. Recently, interest on cell therapy has increased, and the leukodystrophies for which metabolic correction is needed have become first-choice candidates for cell-based clinical trials. In this review, we present and discuss the available cell transplantation therapies in metabolic leukodystrophies including fucosidosis, X-linked adrenoleukodystrophy, metachromatic leukodystrophy, Canavan disease, and Krabbe's disease. We will discuss the latest advances of cell therapy and its pitfalls in this group of disorders, taking into account, among others, the limitations imposed by reduced cell migration in multifocal conditions, the need to achieve corrective enzyme threshold levels, and the growing awareness that not only myelin but also the associated axonopathy needs to be targeted in some leukodystrophies.
Keywords
Introduction
Leukodystrophies are defined as inherited disorders affecting the brain white matter, resulting either in its impaired development (dysmyelination) or in its destruction (demyelination) (4). In some leukodystrophies, the peripheral nervous system (PNS) may also be affected and the presence of abnormal PNS myelin can be observed. In dysmyelination, myelin formation does not proceed appropriately, culminating in myelin with defects in structure and function, which may further increase the susceptibility to myelin damage and loss. On the other hand, demyelination is characterized by a normal synthesis and formation of myelin, which is followed by a decrease or loss of myelin. There are several factors contributing to myelin loss, but as commonly observed in peroxisomal and lysosomal disorders, the accumulation or defect of a given metabolite is generally the major underlying cause.
Currently, there is no cure for leukodystrophies. With an estimated incidence of 1 in 7,500 live births, this group of disorders has a significant societal and economic impact when considering patient care and the massive effort involved in testing and developing therapeutic approaches (8). Due to the frequent recessive nature of the underlying genetic mutations, patients are mostly diagnosed during the symptomatic phase (i.e., after disease onset), with the exception of cases with known family history. Also, severe phenotypic forms of leukodystrophy generally lead to progressive and extremely fast degeneration, being the window of action for a therapeutic intervention very narrow. Additionally, due to the fact that some leukodystrophies can affect both the central nervous system (CNS) and PNS, effective treatments may need to target both systems (Table 1). Moreover, the blood–brain barrier (BBB) may represent an additional challenge to devise therapies that will effectively target the brain and spinal cord parenchyma and correct the neuropathology. In addition, as leukodystrophies are a very heterogeneous group of disorders, specific treatments need to be designed according to the characteristics of each disease.
Metabolic Leukodystrophies in Which Cell Transplantation Has Been Used as a Therapeutic Option
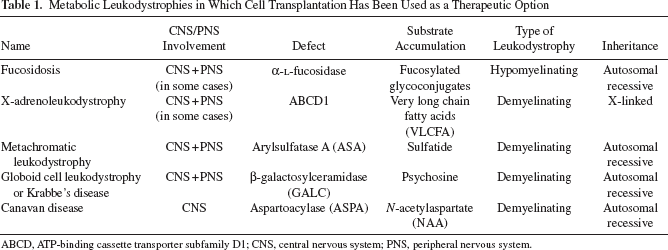
ABCD, ATP-binding cassette transporter subfamily D1; CNS, central nervous system; PNS, peripheral nervous system.
The usage of cellular therapy (e.g., transplantation of stem cells or transplantation of differentiated cells) has been gaining momentum and relevance, as this approach may have a broad therapeutic action. Upon delivery, transplanted cells may act at different synergistic levels, which include (i) acting as a continuous source of the missing enzyme (either naturally secreted or genetically engineered to be a vehicle for protein delivery); (ii) replacing affected/lost host cells; and/or (iii) providing the affected tissues with supporting factors that may delay disease progression and promote recovery. Another crucial aspect on the use of cell transplantation in leukodystrophies is their potential to migrate to affected tissues and provide a global therapeutic strategy. In this respect, different cell sources and transducing vectors have been tested to enable a more widespread and safer delivery, including to the barrier-protected brain. The ability of the allogeneic transplanted cells to serve as a source of normal protein/enzyme may function in two different paradigms. The first paradigm resembles the enzyme replacement therapy (ERT), which has been used with success in several lysosomal disorders (3, 13, 99). The transplanted cells may secrete the active enzyme that can be taken up by neighboring cells through mannose-6-phosphate receptor-mediated endocytosis (30, 113), leading to cross-correction of multiple neighboring cells. In the second paradigm, the transplanted cells, having normal enzymatic activities, can take over the synthesis and/or degradation of the metabolite, which is abnormal in the tissue parenchyma, and may locally provide normalization of metabolite levels. The ability of the allogeneic transplanted cells to replace affected/ lost host cells is still a controversial topic (95, 110, 117), since it involves the transplantation of undifferentiated or progenitor cells, which upon delivery can differentiate or transdifferentiate and repopulate a given tissue. For some leukodystrophies, transplantation of hematopoietic stem cells may have beneficial effects, since donor-derived cells can enter nervous tissues and differentiate into microglia and astrocytes (18, 66). Donor-derived monocytes that enter the nervous tissue and differentiate into microglia can exert multiple beneficial functions, including repopulation of the tissue, secretion of the defective enzyme or of supportive trophic factors for neurons and glia, provision of localized normalization of metabolic functions, and mediation of anti- or proinflammatory actions (101). Another possible therapeutic cell source in leukodystrophies is myelin-forming precursors/cells, as they can both produce the missing enzyme and replace the host-damaged cells. For that purpose, neural stem/progenitor cells or oligodendrocyte progenitors have been used to target the CNS, and Schwann cells have been used to target the PNS. Embryonic stem cells either uncommitted or committed into a neural progenitor stage have also been considered as a possible cell source for replacing the use of glial progenitor cells to overcome their scarce availability. Finally, the usage of bone marrow-derived mesenchymal stromal cells (BM-MSCs) has recently been suggested as an alternative therapeutic cell source for leukodystrophies. Due to their well-known secretory properties, BM-MSCs can act not only by providing the defective protein but also by supporting neighboring cells through the secretion of a wide array of neurotrophic factors and chemokines (12, 15, 62, 106). Additionally, they can also act by modulating inflammation (112, 124). BM-MSCs are easy to harvest and expand without major ethical problems, lack tumorigenic potential (14), and have a strong ability to home to injured tissues after being systemically injected (41). Moreover, due to their low immunogenicity, they can be used in allogeneic transplants without matching donors and recipients (1). Cell transplantation using stem/progenitor cells has allowed some restoration of the metabolic defect. However, the usual low migration rate, as well as the low expression levels of the missing protein, entails a decreased efficiency in terms of therapeutic outcome.
Therapeutic interventions in some leukodystrophies using cellular therapies have yielded sufficient beneficial outcomes to warrant further successful developments. It is understandable that in most cases, regardless of beneficial outcomes, the transplantation may not be sufficient to provide the broad rescue needed in terms of metabolism and tissue pathology. From this perspective, there is the need to test and develop additional therapeutic strategies specifically aimed at rescuing a given aspect of the disease. In this review, we will discuss the most recent studies using cell transplantation as a therapeutic solution for metabolic leukodystrophies (summarized in Table 2). The latest advances, as well as the major breakthroughs in surpassing therapeutic challenges, will then be debated.
Summary of Cell Therapy-Based Therapeutic Approaches in Metabolic Leukodystrophies

Cell source applied to humans or animal models, delivery route, and specific targets and outcomes are indicated for each metabolic leukodystrophy. ASA, arylsufatase A; BM-MSCs, bone marrow-derived mesenchymal stromal cells; CD, Canavan disease; CNS, central nervous system; ESGPs, glia precursors derived from murine embryonic stem cells; GALC, β-galactosylceramidase; GLD, globoid cell leukodystrophy; IC, intracerebral; ICV, intracerebroventricular; IP, intraperitoneal; IV, intravenous; HSCs, hematopoietic stem cells; KD, Krabbe's disease; MLD, metachromatic leukodystrophy; NCV, nerve conduction velocity; ND, not determined; NPCs, neural progenitor cells; NSCs, neural stem cells; OLs, oligodendrocytes; OLPs, oligodendrocyte progenitor cells; PNS, peripheral nervous system; UCB, umbilical cord blood; VLCFA, very long chain fatty acids; X-ALD, X-linked adrenoleukodystrophy.
Leukodystrophies Targeted with Cell Transplantation
Fucosidosis
Fucosidosis is an autosomal recessive lysosomal storage disease caused by defective α-l-fucosidase due to mutations in the FUCA1 gene (123). Deficiency in α-fucosidase leads to the accumulation of fucosylated glycoconjugates (e.g., glycoproteins and glycolipids) in tissues and urine (81, 111). The clinical presentation of fucosidosis consists of a severe neurodegeneration with rapid and progressive mental and motor deterioration (122). Similar to other lysosomal disorders, the disease affects nonnervous tissues, with patients presenting facial dysmorphia, gingival hypertrophy, angiokeratoma, visceromegaly, ocular abnormalities, and hearing loss. In the CNS, the main magnetic resonance imaging (MRI) finding is a generalized hypomyelination of white matter tracks (74, 122).
The canine and, more recently, the feline models for fucosidosis have been important tools to study the disease and evaluate therapeutic interventions (2, 42). In English Springer spaniel dogs with fucosidosis, preceding clinical signs, histological analysis of brains revealed vacuolation of neurons, astrocytes, and microglia. The hypomyelination seen in human patients is reflected in the canine model by a generalized decrease in the expression of relevant myelination genes (26, 52). Bone marrow transplantation in dogs with fucosidosis is able to increase fucosidase activity in plasma, leukocytes, and visceral tissues (107). Importantly in the context of a leukodystrophy with a neuronal component, neural tissues also showed increased enzymatic activity. The improvements in the pathology of the PNS and CNS are clear indicators of the effectiveness of bone marrow transplantation in the treatment of fucosidosis. Nevertheless and because the canine model resembles closely the human disease, it has been shown that therapeutic interventions after the appearance of clinical symptoms are ineffective (24, 109). A gene therapy trial has also been tested in the canine model using retroviral gene transfer into autologous hematopoietic stem cells (23). Despite the lack of engraftment, given the recent advances in vector design, transduction, and in vitro manipulation of bone marrow cells, improved strategies should be pursued to delineate the best approach for defining a cellular therapy for fucosidosis. ERT has also been evaluated in fucosidosis dogs (51). Intracisternal delivery of recombinant canine α-l-fucosidase was able to reduce the accumulation of fucosylated glycoconjugates. Given these results and that an ERT can be initiated before the development of symptoms, combinatorial approaches including ERT and transplantation may prove to be more beneficial than a single therapeutic intervention.
Bone marrow transplantation has also been performed in fucosidosis patients (74, 116). After successful grafting, a progressive increase in α-l-fucosidase levels was observed in lymphocytes, plasma, and cerebrospinal fluid. Concomitantly with the increase in enzymatic activity, an improvement in myelination was observed. MRI examination for up to 46 months after transplantation showed a near normalization of myelination, although there was a decrease in psychomotor functions. The caveat behind the knowledge gathered from the human and canine trials resides in the time of intervention. In disorders like fucosidosis, the rapid decline after the appearance of first symptoms may hinder the benefits of bone marrow transplantation, since the cellular alterations and tissue pathology cannot be rescued by donor-derived monocytes or differentiated cells. Nevertheless, these studies have highlighted the need to improve the diagnosis and functional assessment in this disease to increase the time frame available to consider cellular therapy.
X-Linked Adrenoleukodystrophy
X-linked adrenoleukodystrophy is a peroxisomal disorder characterized by cerebral demyelination, adrenal insufficiency, and progressive neurological deterioration. X-linked adrenoleukodystrophy is caused by a defect in the ABCD1 gene, coding for the peroxisomal membrane protein ALD (ALDP), which belongs to the ATP-binding cassette protein family (21, 79, 80). Its metabolic hallmark is the accumulation of very long chain fatty acids (VLCFA) in tissues and plasma due to impaired transport and β-oxidation of these fatty acids in peroxisomes (37, 43, 44, 115, 118). X-linked adrenoleukodystrophy is an inflammatory demyelinating disorder (22, 87) that in its most severe form affects children with a rapid and progressive demyelination. The inflammatory demyelination also affects neurons and their axons, although axonopathy is the prevalent presentation of adrenomyeloneuropathy (86), an adult presentation of X-linked adrenoleukodystrophy.
Removal of accumulated VLCFA may be a possible treatment for adrenoleukodystrophy. Treatment with Lorenzo's oil (a mixture of glyceroltrioleate and glyceroltrierucate), which normalizes plasma VLCFA concentrations, has no effect in patients with established neurological deficits, as brain levels of VLCFA are unchanged by the treatment (114). 4-Phenylbutyrate and fenofibrate are compounds known for their ability to reduce VLCFA levels in cultured cells and in Abcd1 knockout mice, due to enhanced peroxisomal proliferation and upregulation of genes involved in β-oxidation and fatty acid transport (125). However, the efficacy and usefulness of these compounds for X-linked adrenoleukodystrophy patients has not been established so far.
Hematopoietic stem cell transplantation (HSCT) is accepted as an effective therapy for the early stages of X-linked adrenoleukodystrophy. A follow-up of patients with X-linked adrenoleukodystrophy showed that only boys with early-stage disease benefit from HSCT (84). The neurological benefits of HSCT in X-linked adrenoleukodystrophy are mediated by the replacement of brain microglial cells derived from donor bone marrow myeloid cells (89). Other than the time of treatment, HSCT is limited by the existence of human leukocyte antigen-matched donors (10) and carries a considerable risk of mortality associated with severe graft-versus-host disease (GVHD) and prolonged immune deficiency (9). Animal models for adrenoleukodystrophy are limited to the Abcd1 knockout mice (28, 67, 91) and the Drosophila recessive mutant “bubblegum” exhibiting adult neurodegeneration (75). The Abcd1 knockout mice generated by target disruption (28, 67, 91) present the expected motor and biochemical impairments early on, but neurological symptoms appear only at a later stage. To clarify the mechanisms of HSCT, intravenous injection was performed in Abcd1 mutant mice (125). Treatment led to a significant reduction of VLCFA accumulation in the spleen and lung after transplantation, but not in the brain and spinal cord. This correlated with a higher engraftment of transplanted cells in the spleen and lung, along with detection of the ALD protein, and little engraftment in the CNS with no detection of ALD protein (125).
In another study, beneficial effects in the CNS are described for two patients with X-linked adrenoleukodystrophy (10). Autologous bone marrow-derived CD34+ cells were isolated from patients, genetically corrected ex vivo with a lentiviral vector encoding the normal ABCD1 gene, and then reinfused into the patients after myeloablative treatment. The cells grafted and expressed ALD, stabilizing the levels of the protein to 10% of the normal values at 30 months after transplant in one patient and to 15% at 24 months in the other patient. VLCFA levels were reduced by 20% and 28% in peripheral blood mononuclear cells from patient 1 and patient 2 at 24 and 20 months posttransplant, respectively. In both patients, treatment led to a halt in progression of cerebral demyelination and cognitive decline. As the autologous hematopoietic stem cells were transduced to express functional ALD protein, a smaller amount of corrected cells was needed to obtain comparable neurological benefits to allogeneic HSCT, this last one being associated with a higher morbidity and mortality risk (10). Similarly, the transplantation of lentiviral transduced murine Abcd1 mutant Sca-1+ cells, a functional equivalent of CD34+ cells in humans, into Abcd1 mutant mice resulted in the replacement of 20–25% of brain microglial cells expressing the ALD protein 12 months after transplantation (9) (Fig. 1).

Repopulation of brain microglia from adrenoleukodystrophy mice by lentivirally transduced bone marrow derived cells. The ratio of the ATP binding cassette transporter subfamily D (ALD) protein- and ionized calcium binding adaptor molecule 1 (Iba-1)-positive cells to the total number of Iba-1-positive cells increases with time after transplant of lentiviral transduced stem cell antigen-1-positive (Sca-1+) cells in adrenoleukodystrophy mice. Insets show microglial cells expressing Iba-1 in green and lentiviral encoded human ALD protein in red with a punctate aspect. From (10), reprinted with permission from AAAS.
In summary, systemic delivery of hematopoietic stem cells enables a satisfactory engraftment in peripheral tissues. However, CNS engraftment is achieved to a lesser extent, correlating with minor metabolic correction in the nervous tissue. It is possible that this limitation might be surpassed by genetically modifying the stem cells to produce higher levels of the ALD protein. It is also important to note that the development of new, accurate models of disease is important to the development of therapies. Recently, X-linked adrenoleukodystrophyinduced pluripotent stem cells, to serve as a cellular model of the disease, have been developed (39). In addition, the double Abcd1: Abcd2 mutant mice (25, 90) have been shown to develop some key pathological features of X-linked adrenoleukodystrophy and of its adult variant adrenomyeloneuropathy and have been important tools in the development and evaluation of therapies aimed at preventing oxidative damage caused by VLCFA accumulation (65). As in many other leukodystrophies, the multifaceted tissue pathology and secondary metabolic and cellular abnormalities may only be targeted with combined therapies. Nevertheless, one must not deviate significantly from the primary cause of the disease, and rescuing or preventing the accumulation of VLCFA in X-linked adrenoleukodystrophy should also still be a prime target for future interventions.
Metachromatic Leukodystrophy
Metachromatic leukodystrophy is a storage disorder that is caused by the inherited deficiency of the lysosomal enzyme arylsulfatase A (ASA). ASA catalyzes the desulfation of cerebroside 3-sulfate (sulfatide), an acidic sphingolipid mainly present in myelin sheaths (50). Deficiency of ASA causes the accumulation and massive intralysosomal deposition of this sphingolipid. Metachromatic leukodystrophy is further characterized by a progressive and widespread loss of myelinating cells leading to severe dysfunction of the nervous system (72). Neurological findings may include spasticity, neuropathy, dementia, and seizures. Importantly, the time of onset and severity correlate with residual ASA activity (85).
The only available animal model for metachromatic leukodystrophy is the ASA null mice (33). ASA knockout mice display progressive sulfatide storage throughout the CNS, mild loss of myelin, and develop behavioral deficits in the adulthood, being a murine model for late infantile/early adult forms of human metachromatic leukodystrophy (32). At approximately 5 months of age, metachromatic leukodystrophy mice display conduction abnormalities in the CNS and PNS, impaired motor coordination and learning, and sulfatide storage in the nervous system (5). In the last decade, the potential of using cell transplantation as a therapy for metachromatic leukodystrophy has been explored in a number of studies, after the initial results showing that HSCT performed in late-onset presymptomatic patients could halt disease progression. Nevertheless, HSCT is only suitable for patients with mild nervous system involvement and with results only seen in the CNS and not in the PNS (4).
In metachromatic leukodystrophy, after HSCT, neurological abnormalities may persist, and patients might benefit from a combinatorial therapy (48). Although the bone marrow contains both hematopoietic and nonhematopoietic progenitors, including BM-MSCs, allogeneic HSCT does not result in replacement of BM-MSCs, which remain of host origin (49). In line with this, in one clinical trial, transplantation of BM-MSCs was performed in metachromatic leukodystrophy patients that previously underwent HSCT (48). In four of six patients in whom BM-MSCs have been transplanted after receiving allogeneic hematopoietic stem cells, significant improvements in nerve conduction velocities were observed only after the BM-MSC infusions (48).
To evaluate further therapeutic strategies in metachromatic leukodystrophy, the long-term outcome of transplanting ASA knockout mice with retrovirally transduced hematopoietic stem cells expressing high levels of ASA was assessed (72). Analyses of 1-year-old animals intravenously transplanted at 5–6 weeks of age showed a reduction of storage material in the liver and kidney. In the CNS, there was a therapy-mediated improvement of sulfatide catabolism (assessed as galactosylceramide/ sulfatide ratio) but there was not a significant reduction of sulfatide storage (Fig. 2). A delay in neuronal loss was not observed after gene transfer. Moreover, although there was a significant increase in myelinated fiber diameter in the PNS, the same was not observed in the CNS. After treatment, a slight improvement of neuromotor abilities was detected. The threshold level of enzyme activity needed for the correction of the metabolic defect was determined and was found to be higher when compared to other lipid storage diseases (72). In the brain, which is less accessible to bone marrow-derived cells than visceral tissues, this threshold level was not attained, thus explaining the lack of improvement.

Sulfatide storage in (a, b) liver, (c, d) kidney, and (e, f) brain of mock-treated (indicated as –; a, c, e) and treated (indicated as +; b, d, f) ASA-deficient mice 10–11 months of age. Mock-treated arylsulfatase A (ASA)-deficient mice were transplanted with nontransduced ASA-deficient hematopoietic stem cells (HSCs), and treated ASA-deficient mice received retrovirally transduced bone marrow stem cells overexpressing human ASA. Intralysosomal sulfatide granules are stained with Alcian blue. A decline of storage is visible in liver (the arrow in b indicates the bile duct epithelium) and kidney, but not in brain. Adapted from (72) with permission from Macmillan Publishers Ltd. (Gene Therapy).
A similar study used the same cell type but expressing the ASA gene in a lentivirus instead of a retroviral vector. Full reconstitution of enzyme activity in the hematopoietic system of metachromatic leukodystrophy mice was achieved. In contrast to the previous study, improvements were noted both in the CNS and PNS. Transgene-expressing progeny of long-term repopulating hematopoietic stem cells were able to repopulate the CNS microglia and PNS macrophages. Treatment prevented the development of motor conduction impairment, learning and coordination deficits, as well as neuropathological abnormalities typical of the disease (6). In contrast, when animals were transplanted with wild-type hematopoietic stem cells without ASA overexpression, abundant metachromatic deposits in the disease target areas and demyelinated fibers in the sciatic nerve were still observed. These results highlight the crucial role of enzyme overexpression, showing a direct correlation between the level of available enzyme and efficacy of phenotypic correction (6). The results of this study are promising and justify the investment in future studies to further address the safety of lentivirus-mediated hematopoietic stem cell gene therapy.
Aiming at developing better therapeutic solutions, cell types other than hematopoietic stem cells have been used in metachromatic leukodystrophy, including embryonic stem cells and oligodendrocyte progenitors, and distinct outcomes have been obtained. Glial precursors derived from murine embryonic stem cells were stably transfected with a plasmid containing the human ASA cDNA, leading up to a 30-fold increase in ASA activity. Cells were transplanted in the brains of newborn ASA-deficient mice as limited results were obtained when 5-week-old mice were used. The majority of the donor cells adopted an astrocytic phenotype with an approximately 50% reduction of sulfatide storage granules around individual ASA-positive cells (45). However, despite the increased ASA levels, the delivery radius in vivo remained small. Hence, clinical strategies for stem cell-based enzyme delivery may have to include techniques that enhance both the enzyme expression and the migration potential of transplanted cells. Oligodendrocyte progenitors migrated extensively within the metachromatic leukodystrophy neonatal brain after injection of the cells in the left ventricle (32). Interestingly, cells migrated rapidly within the metachromatic leukodystrophy brain parenchyma and less within the wild-type tissue. Also, grafted cells remained in metachromatic leukodystrophy brains for a longer period of time than in transplanted age-matched healthy controls. Grafted progenitors differentiated into mature myelinating oligodendrocytes and integrated in myelinated areas of the metachromatic leukodystrophy brain. Typical features of the disease, such as astrocytosis, microglia activation, and macrophage infiltration, were reduced by the treatment. Electrophysiological and motor learning impairments were prevented in transplanted metachromatic leukodystrophy mice, as evaluated in 1-year-old mice. There was a reduction of 20–50% of sulfatide deposits in the metachromatic leukodystrophy CNS that correlated with ~31% reconstitution of the normal ASA activity after neonatal transplants of oligodendrocyte progenitors (32). In another study, the same group showed that although multipotent neural stem cells differentiated into astrocytes, they were still able to exert beneficial therapeutic effects. Contrary to what was observed for oligodendrocyte progenitors, sulfatides had a deleterious effect on the capacity of brain implanted multipotent stem cells to form oligodendrocytes. It is important to underline that the reduced neurodegeneration was accompanied by increased ASA activity and reduced sulfatide accumulation in transplanted metachromatic leukodystrophy brains, indicating a correlation between the correction of the enzyme deficit, sulfatide accumulation, and neuroprotection (31).
Another approach that has been explored took advantage of the capacity of homeobox B4 (HoxB4) to regulate hematopoietic stem cell proliferation (98) by transplanting hematopoietic stem cells overexpressing HoxB4 in metachromatic leukodystrophy mice. Transdifferentiation into oligodendrocytes, although at a low rate, was reported as well as a better motor performance (77). More recently, umbilical cord blood (UCB) transplantation (UCBT) has been considered as a potential better option than bone marrow-derived hematopoietic stem cells, as stored and cataloged UCB can be rapidly identified and transplanted, producing a shorter period between diagnosis and transplantation, which is an important issue to consider in neurodegenerative diseases. A study of three siblings with metachromatic leukodystrophy that underwent UCBT at different stages of disease has been performed (85); for the two younger siblings, UCBT has stopped disease progression. Compared to bone marrow transplantation, UCBT has the advantage of being quickly available, presenting a lower risk of GVHD together with higher compatibility rates and less risk of infections (85).
Overall, although therapeutic advances have been made in metachromatic leukodystrophy, there is still the need to optimize procedures that will have a simultaneous impact in the CNS and PNS, tackling limitations of cell migration and enzyme threshold levels. In this sense, metachromatic leukodystrophy is a good candidate to explore combinatorial therapies that might achieve a broad range of action with sustained results.
Krabbe's Disease or Globoid Cell Leukodystrophy
Krabbe's disease is an autosomal recessive disorder caused by mutations in the lysosomal enzyme of β-galactosylceramidase or galactocerebroside β-galactosidase (GALC) (119). Accumulation of one of the GALC substrates, the toxic lipid raft-associated sphingolipid psychosine, causes apoptosis of myelin-forming oligodendrocytes and Schwann cells, leading to severe demyelination of both CNS and PNS (38, 46, 121, 126). Besides myelin impairment, Krabbe's disease is characterized by astrocytosis as well as by the accumulation of macrophages in the PNS or microglia-derived cells—the globoid cells—in the CNS, both containing inclusions of galactosylceramide, the other GALC substrate (40). This generates a strong inflammatory environment and the characteristic presence of globoid cells, thus the alternative name of this disease, globoid cell leukodystrophy.
Krabbe's disease has several forms according to the age of onset. The most prevalent phenotype is the classic early infantile form, which consists of approximately 90% of all cases. In this subtype, children develop the first symptoms before 6 months of age and usually die before they complete 2 years. The symptoms are numerous but may not significantly vary between the early variants of disease and may include hyperirritability, arrest of motor and mental development followed by regression, spastic paraparesis, cerebellar ataxia, optic atrophy with consequent visual deterioration, dystonia, epileptic seizures, hyperpyrexia, or psychosis; respiratory infections may also occur and are sometimes the cause of death (55). Besides the early infantile form, late phenotypes also exist and are clinically very heterogeneous. Peripheral neuropathy has additionally been reported to occur in Krabbe's disease patients, especially in the infantile form, occasionally showing an extremely rapid development (54).
The Twitcher mouse is a natural occurring murine model for human Krabbe's disease (47, 96) where psychosine accumulation is also considered to be cytotoxic for myelin-forming cells (78), causing degeneration of oligodendrocytes and Schwann cells (104). In Twitcher mice, symptomatology presents high similarity with humans: mice develop tremors (twitching) at around postnatal days (PND) 18–20 and subsequently develop muscle weakness, severe weight loss, and eventual paralysis of the hind limbs, and they hardly survive beyond 45 PND (105). Other animal models such as cats, dogs (27), and primates (69) also exist, although they are used less frequently than the murine models.
Currently, there is no cure for Krabbe's disease. Substrate reduction therapy, which in this particular context consists in the administration of substances designed to reduce the synthesis of the accumulating glycosphingolipids, has been suggested as one possible treatment, especially for late-onset forms (7). Even though treated Twitcher mice lived longer and had a slower clinical course of disease progression, they continued to worsen and died earlier than normal controls (61, 82). Another approach already tested in Twitcher mice is ERT: animals received intraperitoneal injections of recombinant GALC periodically (either once per week or once every other day) beginning at PND 10 or 20 (58). GALC uptake was seen in multiple tissues, including a small amount in the brain (58). Peripheral macrophages were probably the cells responsible for delivery of exogenous GALC to the CNS. Nevertheless, ERT could not avoid the quick decline of the animals. Later, a single intracerebroventricular administration of GALC aiming at directly delivering the enzyme to the Twitcher brain was performed (59). Despite an increase in life span and a reduction in psychosine substrate accumulation, no effective treatment was achieved, as the animals only survived up to the age of 51 days.
Successful GALC delivery approaches have been described for Krabbe's disease, with widespread enzymatic correction of the CNS and a decrease in both astroglia and microglia (57). Using intracerebral injections of either adeno-associated virus or lentivirus encoding the mouse GALC cDNA, high levels for GALC activity were obtained with significant correction of the pathology in brains of Twitcher mice (57, 64). However, with this approach, only a modest increase in life span, body weight, and in the success of behavioral tests has been obtained, even when treating newborn mice, before the disease onset. At terminal stages, demyelination and globoid cells were present at the same extent as in untreated animals (64). These data suggest that the simple increase in enzyme activity is not the key to solve this disease. Still, similarly to other leukodystrophies, in Krabbe's disease, gene therapy has also been used as a way of producing transduced cells with higher GALC activity, allowing an increased amount of enzyme to be delivered. In vitro, the exogenous donor-derived enzyme is efficaciously taken up by neighboring Twitcher cells (fibroblasts or glial cells) to their lysosomal fraction, promoting their effective cross-correction (68, 92).
Presently, the only available therapy for Krabbe's disease patients is HSCT (either bone marrow or UCB-derived), which is efficient in presymptomatic Krabbe's disease newborns diagnosed because of associated family history (20). Time of transplantation is then an extremely important issue in Krabbe's disease. The prognosis of UCB-transplanted Krabbe's disease patients strongly depends on the stage of disease at which transplantation is performed (19). If performed after the onset of symptoms, patients show only mild improvements, and the disease course is not changed (20). Additionally, a strong impairment of the PNS is still found in transplanted patients (20, 88, 100). Emphasizing the need of correcting the peripheral nerve pathology, studies in transplanted Twitcher mice have shown similar disappointing results in the PNS (36, 53): Despite that HSCT consistently alleviates to some extent the neurological symptoms in Krabbe's disease and increases the life span of transplanted mice (34, 56), just a slight improvement of the PNS pathology is obtained (35, 53).
Since August 2006, in New York State, Krabbe's disease has been added to the group of disorders screened in newborn infants (16). Despite that performing this type of screening for the detection of infants that bear the infantile form of the disease may be advantageous, the present suboptimal results with hematopoietic stem cell transplants are discouraging. Additionally, therapy in patients with later onset forms of the disease is discussable, as this disease type shows a lot of variability in the disease expression phenotypes, with unpredictable progression. As such, the expansion of the newborn screening to other states is presently a controversial issue (97). In summary, although HSCT improves survival and promotes some mild neurological benefits, it does not cure Krabbe's disease patients. Hence, additional therapies, probably combinatorial therapies to be administered together with HSCT, must be developed.
As Krabbe's disease is a disorder specifically affecting the nervous system, the primary obvious cell source for therapeutic approaches is neural stem cells. Moreover, both murine and human neural stem cells are resistant to psychosine (108), as they can engraft and distribute throughout demyelinating areas of the Twitcher mouse brain, even at juvenile ages (83, 108, 127). As neural stem cells can differentiate after grafting (127) and survive to the toxic environment of psychosine, it is possible that cell replacement takes place (108). GALC-expressing engineered neural stem cells promote myelination and increase the life span of Twitcher mice up to two- to threefold (108). Although a promising cell source, the development of neural stem cell-based therapeutic approaches is limited by their availability.
Recently, the interest in BM-MSCs has been increasing in cell therapies (76, 94). In an initial study, promising results were obtained by intracranial administration of murine BM-MSCs into neonatal Twitcher mice (94). An increase in life span, body weight, and motor function was observed in transplanted mice, which was attributed to the anti-inflammatory properties of BM-MSCs. In a study from our group, an enhanced green fluorescent protein-positive (GFP+) cell line of BM-MSCs, immortalized with telomerase reverse transcriptase, was systemically delivered into Twitcher mice to test whether this cell type would ameliorate the PNS pathology (76). BM-MSCs grafted the sciatic nerve (Fig. 3) and promoted a diminished neuropathology, as determined by a significant increase in Schwann cell precursors and axonal number. Curiously, axonal degeneration arising independently of myelin loss has been recently proposed to be the cause for the lack of efficacy of available treatments, essentially aiming at correcting the myelin defects (11, 29). Therefore, BM-MSCs may represent a valuable add-on option to current therapies aiming at promoting neuroprotection in leukodystrophies, specifically in the PNS.

Bone marrow mesenchymal stromal cells (BM-MSCs) grafted into Twitcher sciatic nerves retain an undifferentiated phenotype. Anti-Sca-1 (left) colocalizes with anti-enhanced green fluorescent protein (EGFP) (right; a double-positive cell is highlighted with an arrow) in Twitcher sciatic nerves 14 days posttransplantation with BM-MSCs. Counterstaining was performed with hematoxylin.
Krabbe's disease is a complex disorder, as its pathology involves psychosine and galactosylceramide accumulation with consequent myelin loss, cell death by apoptosis, macrophage invasion, and inflammatory response, thus increasing the challenge at simultaneously addressing these different disease mechanisms (17). Recently, it was proposed that psychosine persists in brain lipid rafts after in vivo enzyme replacement, which is another limiting factor for the current therapies (120). As such, a combined approach is probably required. Lately, some combinatory treatments for Krabbe's disease have been proposed. Most of the dual approaches use HSCT and the induction of increased expression of GALC by gene therapy (29, 63, 93). In these studies, combined therapies had a synergistic effect on the life span of Twitcher mice, and GALC activity was restored. In some cases, improved locomotor and behavioral performances, significant preservation of myelin, and reduction of macrophage infiltration and astrocytosis were achieved; nonetheless, at the end, all treated Twitcher mice died with symptoms of progressive neuronal degeneration (29, 63, 93). Additional studies focusing on disease mechanisms will be very helpful in understanding the failure of current Krabbe's disease therapies.
Canavan Disease
Canavan disease is an autosomal recessive leukodystrophy caused by a defect in aspartoacylase (ASPA). ASPA deficiency leads to accumulation of N-acetylaspartate (NAA) in the brain (70). This enzyme is synthesized by oligodendrocytes that are lost in Canavan disease. The clinical symptoms include megalencephaly, hypotonia, mental retardation, and early death (103). Treatment for Canavan disease is symptomatic, and few studies have been performed to devise viable therapies. Gene therapy by ASPA gene transfer using a nonviral delivery system in conjunction with adeno-associated virus-based plasmids into the brain of two children with Canavan disease reduced NAA accumulation, but long-term follow-up is lacking (60).
The knockout mouse model for Canavan disease shows similar abnormalities to those observed in patients including ASPA deficiency, accumulation of NAA, and spongy degeneration of the brain (71). This year, a newly engineered model of Canavan disease, the ASPA-LacZ knockin mouse, was described (73). As shown for other leukodystrophies characterized by a defective enzyme product, the use of stem cells as a vehicle for enzyme delivery has been tested in the Canavan disease mouse model. Retrovirally transfected neurospheres overexpressing ASPA were implanted into the brain in the Canavan disease mouse (103). Neural progenitor cells survived, migrated, and differentiated in vivo into oligodendrocyte progenitor cells in the juvenile knockout mouse brain. Positive staining of the implanted cells suggested that neural progenitor cells differentiated into oligodendrocytes (Fig. 4), potentially leading to remyelination (103). This is of great importance as loss of ASPA synthesizing cells, oligodendrocytes, is one of the key events in Canavan disease. The transplanted neural progenitor cells led to measurable ASPA activity in the mouse brain up to 1 month posttransplantation, the maximum time period studied (103). As in Canavan disease fibrous astrocytes, localized within the white matter, are also affected showing astrocytic swelling, the ability of transplanted neural progenitor cells to differentiate into astrocytes was tested. The presence of glial fibrillary acidic protein (GFAP)-positive transplanted cells in the Canavan disease mouse brain suggested that neural progenitor cells can also differentiate into astrocytes and can therefore be used to replace lost GFAP-positive cells in the Canavan disease brain. Together, these results suggest that cell therapy is a promising option for Canavan disease.

Transplanted neural progenitor cells differentiate into oligodendrocyte progenitor cells in the Canavan disease mouse brain. Neural progenitor cells stained with the oligodendrocyte progenitor marker, NG2, showed positively stained cells (green). Bromodeoxyuridine (BrdU)-positive cells (red). Adapted from (103) with permission from Elsevier.
Conclusions
Presently, stem cell transplantation represents a step forward in the treatment of leukodystrophies as it enables a multilevel mode of action where transplanted cells do not only provide the missing protein but may also replace damaged cells, secrete supportive factors, and play an immunoregulatory role. However, some challenges remain, and new approaches are urgently needed to design successful clinical therapies.
As discussed in this review, in some of these disorders such as metachromatic leukodystrophy, Krabbe's disease, and X-linked adrenoleukodystrophy, HSCT and UCBT are able to stabilize neurodegeneration, although with some limited efficacy, as they have to be applied at the asymptomatic stage or early in disease progression so that a beneficial outcome is achieved. On the other hand, generally, this therapeutic approach does not correct the PNS impairment. As such, alternative stem cell sources to be used in combination with hematopoietic stem cells have been explored in animal models. Their transference to the clinics awaits further tests of efficacy and safety.
When compared to hematopoietic stem cells or mesenchymal stromal cells, neural stem cells and oligodendrocyte progenitors have the advantage of being able to differentiate into neural cells, replacing the damaged glia, while secreting the enzyme that is missing to the neighboring cells. Some studies in metachromatic leukodystrophy and Krabbe's disease suggested that these cell types may be more beneficial than hematopoietic stem cells. Nonetheless, neural stem cells and oligodendrocyte progenitors have the obvious limitation of still being difficult to obtain in sufficient large numbers that would allow transplantation in humans. In the case of embryonic cells such as glial precursor-derived embryonic stem cells, ethical issues are an additional limitation to their use for clinical purposes in a near future. Irrespective of the cell source, in several leukodystrophies, genetically engineered stem cells allowing both increased enzyme expression rate and migration are most probably the route to be followed for future clinical applications, as modified cells have been shown to produce increased improvements when compared to the nonmodified counterparts.
With the current knowledge that neuropathology in leukodystrophies is probably more complex than the previously thought myelin-related impairment, the present challenge is to design combinatorial strategies capable of targeting not only myelin-producing cells but also neurons and the immune system. In this context, the usage of bone BM-MSCs as an add-on strategy might prove to be useful: BM-MSCs are capable of rescuing axonopathy in leukodystrophies; they produce a beneficial immunoinflammatory response, enhance hematopoietic stem cell engraftment, decrease GVHD after allogeneic HSCT, and increase the overall performance of HSCT. In diseases where both the PNS and CNS need targeting, cotransplantation of BM-MSCs with other cell types may prove to be beneficial as these successfully target the PNS. Moreover, in the case of HSCT, BM-MSCs create a favorable environment for the action of HSCT and may facilitate the repopulation of microglia that can serve as a cellular vehicle for CNS gene therapy, ensuring rapid and long-term enzyme delivery. It might also be interesting to combine the use of neural stem/progenitor cells with BM-MSCs. With this approach, BM-MSCs may contribute to the generation of an appropriate niche for the engraftment and differentiation of neural stem/progenitor cells, allowing a more successful replacement of damaged cells. Targeting to the injury site might be also potentiated by the synergistic action of both cell types. In fact, one of the most highlighted features of MSCs is their capacity to preferentially home to the sites of injury. In this context, it would be worth exploring if MSCs enhance the migratory and grafting ability of other stem cell types.
In conclusion, in leukodystrophies an effective treatment is still awaited. Currently, cellular therapy is the most promising approach, but the cell sources used so far are not capable of promoting on their own the complete blockage of myelin degeneration or full regeneration after tissue destruction. As such, combined approaches are indispensible to accomplish the full neuropathological rescue in this group of disorders. We suggest that BM-MSCs are strong candidates to be part of such cell transplantation combinatorial approaches for the treatment of leukodystrophies.
Footnotes
Acknowledgments
The work from the author's group was supported by the European Leukodystrophy Association (ELA Foundation No. 2010-042C5A), by FEDER Funds through the Operational Competitiveness Programme–COMPETE and by National Funds through FCT–Fundação para a Ciência e a Tecnologia under the project FCOMP-01-0124-FEDER-015781 (PTDC/SAU-GMG/111761/2009) and FCOMP-01-0124-FEDER-022718 (PEst-C/SAU/LA0002/2011) to M.M.S. C.A.T. was supported by Programa Ciência, funded by POPH-QREN and MCTES, and C.O.M. was supported by FCT (SFRH/BD/29768/2006 and BPD/34811/2007). P.B. was supported by FCOMP-01-0124-FEDER-015970 (PTDC/SAU-ORG/112406/2009) from FCT under the programs FEDER and COMPETE. The authors declare no conflict of interest.