
Abstract
Currently, islet transplantation as a cell therapeutic option for type 1 diabetes occurs via islet injection into the portal vein. Direct contact between islets and blood is a pathophysiological “provocation” that results in the instant blood-mediated inflammatory reaction (IBMIR) and is associated with early islet loss. However, the nature of the various insults on the islets in the blood stream remains mostly unknown. To gain insight into the mechanisms, we utilized a simplified in vitro model in which islets were exposed to blood in different clinically relevant but increasingly challenging, autologous, allogeneic, and xenogeneic combinations. Irrespective of the blood type and species compatibility, islets triggered blood clotting. Islet damage was worse as islet, and blood compatibility diminished, with substantial islet injury after exposure of porcine islets to human blood. Islet damage involved membrane leakage, antibody deposition, complement activation, positive staining for the membrane attack complex, and mitochondrial dysfunction. Islet damage occurred even after exposure to plasma only, and specific complement inactivation and neutralization of IgM substantially prevented islet damage, indicating the importance of humoral immunity. Efficacious measures are needed to reduce this injury, especially in view of a potential clinical use of porcine islets to treat diabetes.
Introduction
Islet transplantation (Tx) can successfully restore glycemic stability in type 1 diabetic patients (42). The fate of isolated islets infused into the portal vein is, however, determined by a number of damaging events, occurring as early as during islet injection. Together, these early factors are estimated to cause a loss of 70% of the transplanted islet mass (29). As a consequence, islets obtained from multiple organ donors become necessary in the majority of the recipients to reach a sufficiently functional islet mass. Roles for cold centrifugation in the purification process (21), low oxygen tension of the portal venous blood (12), an active innate immune system including Kupffer cells (9), and the activation of an instant blood-mediated inflammatory reaction (IBMIR) (4) have been postulated. Nonetheless, the extent of the islet damage and the mechanisms involved early after islet injection in the blood stream are not yet clearly understood.
The availability of human organ donors does not meet the increasing demand for human organs to cure severely debilitating and life-threatening diseases and drastically limits the development of programs of islet Tx. To this aim, the employment of porcine islets for clinical use is currently under investigation and may become a therapeutic option in the near future (18). A better understanding of the events that occur when islets are in contact with blood, particularly in view of the possible use of xenogeneic islets, is therefore urgently needed.
Experimental in vivo studies have shown that an intensive inflammatory reaction occurs when islets are exposed to whole blood. The resulting islet cell damage is reflected by an early nonphysiological peak in circulating C-peptide levels, which we and others have observed (34,45). Some insights into the mechanisms that initiate IBMIR and early islet loss have been provided, such as the involvement of the coagulation and the complement systems (4). Multiple treatment options to circumvent these problems have been proposed and investigated (26,37,41), but promising in vitro results have thus far not been converted into successful prevention of early graft loss in vivo.
The aim of our study was to set up a simplified in vitro test to investigate the events that occur following contact of islets with whole blood or plasma in order to identify approaches that minimize early islet loss in vivo.
Pig or human islets were selectively exposed to autologous, allogeneic, or xenogeneic blood. We found that islets triggered blood clotting regardless of the combinations, whereas islet damage was greater in xenogeneic combinations than that in autologous and allogeneic settings. Prevention of blood clotting (low molecular weight dextran sulfate, LMW-DS) and targeting tissue factor by nacystelyn (NAC) were not sufficient to prevent islet loss, whereas specific complement inhibitor (compstatin) and anti-IgM antibodies efficiently reduced islet damage. These new insights may warrant more efficient protection of pancreatic islets in the peri-Tx phase.
Materials and Methods
Sources of Human and Porcine Islets
Human deceased donor pancreata (n = 11) were obtained from the Center for Organ Recovery and Education (CORE) in Pittsburgh, PA, using standard organ recovery techniques once consent for research use of human tissue was obtained. Islets were isolated using the semiautomated method described by Ricordi et al. (40) with minor modifications (7). The purity of islet preparations was evaluated by dithizone staining (30). The islets were cultured for 1–7 days (37°C, 5% CO2) in CMRL-1066 culture medium (Cellgro Mediatech Herndon, VA) supplemented with 10% heat-inactivated fetal calf serum, 100 units/ml penicillin, 0.1 mg/ml streptomycin, and 2 mmol/L l-glutamine (Life Technologies, Grand Island, NY), until used in the experiments.
Large white crossbred adult sows (n = 11) (Wally Whippo, Enon Valley, PA) and adult pigs transgenic for human CD46 (hCD46) (31), a complement regulatory protein (CRP) [n = 3, 2 of which were on an α1,3-galactosyltransferase gene-knockout (GT-KO) background, therefore lacking expression of the galactose α1,3galactose (Gal) epitope] (Revivicor, Blacksburg, VA), were used as pig islet donors. Methods of recovery of pig pancreata, islet isolation and purification, and evaluation of purity and quality have previously been described (8). All pig procedures were in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Blood Samples
Human blood was drawn from healthy volunteers after informed consent, as approved by the University of Pittsburgh Institutional Review Board (IRB #0608179). Human whole blood was drawn into tubes with 1 mg/ml ethyl enediamine tetraacetic acid (EDTA) for plasma separation or used immediately after being drawn without addition of anticoagulants. Heat inactivation was carried out by incubation of samples at 56°C effectively for 30 min. Pig donor blood was collected into tubes containing 1 mg/ml EDTA. EDTA-anticoagulated human donor blood was received together with the donor pancreas and stored overnight until used in experiments. It was recalcified with calcium chloride (final concentration, 40 mM) to restore its coagulative capacity just before use. Complement activation was only investigated in experiments with freshly drawn blood.
Experimental Design
Approximately 2,500 human or porcine islet equivalents with a purity of 60–80% were resuspended in 1.0 ml of culture medium (CMRL) and placed in 35-mm untreated polystyrene Petri dishes (BD Falcon, Franklin Lakes, NJ). One milliliter of freshly drawn human whole blood or anticoagulated and recalcified donor whole blood was added in autologous, allogeneic, or xenogeneic combinations with the islets. Blood was always ABO blood group-compatible with the islet donor. Plasma alone was added in the amount of 500 μl, and final volume was adjusted accordingly.
Whole blood was added in a 1:1 volume ratio with the islet suspension, which allowed for the sampling of supernatants once the islet-induced fibrinous clot was broken by a pipette tip. This experimental setup reflects the in vivo infusion of islets, resuspended in infusion fluid, into the portal vein. The experiment was performed in an incubator shaker at 37°C and 100 revolutions per minute (rpm). The time until clotting was recorded and compared to controls (1.0 ml of blood and 1.0 ml of medium, but no islets).
Dishes were set up in quadruplicate to allow for supernatant sampling at 5, 15, 30, and 60 min. At sampling, EDTA was added (10 mM final concentration) to prevent further complement activation (35). Supernatants were spun and immediately stored at −70°C until further analysis. Samples for analysis of human or porcine C-peptide were stored with 5% aprotinin (Trasylol, Bayer Pharmaceuticals, West Haven, CT) for protein preservation.
In order to better understand the role of coagulation and complement activation in early islet damage, five independent treatments, relevant to xeno-Tx, were tested. First, LMW-DS (Fluka, Buchs, Switzerland; 1.6 mg/ml) was added. LMW-DS has been used in vitro to prevent islet-induced coagulation and complement activation (26) and in vivo in preclinical islet xeno-Tx models (41,46). Second, NAC (a N-acetyl cysteine derivative, 80 mM, kindly provided by Laboratoires SMB, Brussels, Belgium), recently shown to have an effect on downregulating the expression of tissue factor mRNA (6), was tested. NAC additionally has antioxidant and anticoagulant effects, possibly due to its direct interference with coagulation factors (6,20). Third, we tested the effect of hCD46 expressed on pig islets (31,46). Fourth, we used compstatin, a C3-binding cyclic synthetic peptide that inhibits complement (Tocris Bioscience, Ellisville, MO; 250 μmol/L) (36). Fifth, we added anti-IgM antibody (1:100, Kirkegaard & Perry, Gaithersburg, MD). None of these treatments affected islet viability or function, including C-peptide release. Epinephrine was added (1 μM in three independent experiments) to assess for any possible inhibition of C-peptide release (48).
Qualitative Analysis
Levels of pig and human C-peptide in supernatants were measured by radioimmunoassay (Linco Research, St. Charles, MO) using species-specific antibodies (without crossreactivity between human and pig C-peptide). Enzyme-linked immunosorbent assay (ELISA) was used to determine levels of soluble complement activation products: C4d for the classical pathway, Bb for the alternative pathway, and iC3b for the converged complement pathway (all from Quidel Corporation, San Diego, CA). Islet viability was assessed by fluorescence-activated cell sorting (FACS) analysis using propidium iodide (PI) according to the manufacturer's recommendations (BD Bioscience, San Diego, CA). Prior to analysis, islet cells were dissociated in dissociation buffer (Gibco, Carlsbad, CA), prewarmed at 37°C (25). Islet cells were then incubated for 10 min at 37°C, whereas pipetting was carried out every 3–4 min to ensure cell dissociation. Islet cells were then washed in phosphate-buffered saline (PBS) containing 0.25% bovine serum albumin (BSA, Fraction V, Sigma, St. Louis, MO) and immediately subjected to FACS analysis (23).
Additionally, cell physiological status was assessed by measuring oxygen consumption by pig islet mitochondria. It was measured by a Clark-type oxygen electrode (Oroboros High Resolution Respirometer, Innsbruck, Austria) after a 2-h exposure to xenogeneic plasma (or autologous plasma as control) and a 30-min glucose starvation. Measurements were performed in Krebs buffer supplemented with 0.5% BSA, 20 mM NaHCO3, and 1 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.4. A 2-ml water-jacketed chamber was maintained at 37°C, and the islet solution was constantly stirred with a magnetic stirring bar. After establishing a stable basal respiration with 5 mM glucose, islets were challenged with 20 mM glucose, which resulted in the stimulation of oxygen consumption. Addition of oligomycin, which shuts down mitochondrial ATP production, resulted in a sharp decrease in respiration rate. The residual level of respiration is indicative of the leak through the mitochondrial membrane and, in an indirect way, of the effectiveness of ATP production. Finally, to estimate maximal activity of the respiratory chain, islets were challenged with an uncoupler carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), which dissipates membrane potential and removes all regulatory restrictions from respiratory complexes.
Histology
After 60 min of incubation, islets cells were fixed in 2% paraformaldehyde and frozen for immunofluorescent staining. Cryo-sections were cut and stained using standard immunofluorescent procedures. The primary antibodies were goat anti-human IgG and IgM (cross-reactivity with swine immunoglobulins, 1:1,000, Kirkegaard & Perry), rabbit anti-human C4d (1:20, EMELCA Bioscience, Bergen op Zoom, The Netherlands), mouse anti-human C5b-9 (1:100, Abcam, Cambridge, MA), and rabbit or mouse anti-insulin (1:100, Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies were goat anti-mouse Cy3, goat anti-rabbit Cy3, donkey anti-goat Cy3 (1:500, all from Jackson ImmunoResearch, West Grove, PA), donkey anti-rabbit Alexa 488, and goat anti-rabbit or mouse Alexa 488 (1:500, Molecular Probes, Eugene, OR).
Photographs were taken through a Nikon Eclipse E800 microscope with a Photometrics Cool SNAP digital camera and Nikon C1 confocal system at 40x objective lens and analyzed by MetaMorph imaging analysis software (Molecular Devices, Downingtown, PA).
For each condition, multiple images from at least two independent experiments were taken and analyzed. Representative images were selected.
Statistical Analysis
Continuous variables are expressed as mean ± the standard error of the mean (SEM) and compared using the Student's t test and ANOVA with post hoc corrected comparisons for treated versus untreated conditions. Linear regression was used to analyze if increases over time in supernatant products were significant, and differences in slope were compared. Values of p < 0.05 were considered to indicate a statistically significant difference. All analyses were performed with GraphPad Prism 4 for Macintosh (GraphPad Software, La Jolla, CA).
Results
Islet-Induced Clotting of Whole Blood
In this in vitro test, clotting of freshly drawn, non-anticoagulated human blood and of pig or human blood that was EDTA-anticoagulated and subsequently recalcified occurred in 30–40 min on average and did not significantly differ (p > 0.05). Addition of islets to the blood rapidly induced total clotting, indicating activation of coagulation and platelet consumption, in human autologous (5:26 ± 0:29 min, p < 0.01 vs. control, thus blood only), pig autologous (4:06 ± 0:21 min, p < 0.001 vs. control), human allogeneic (3:13 ± 0:27 min, p < 0.001 vs. control), and pig-to-human xenogeneic combinations (3:54 ± 0:32 min, p < 0.001 vs. control) (Fig. 1A). Clotting time did not significantly differ when comparing autologous, allogeneic, and xenogeneic settings (p > 0.05).
(A) Islets induced complete clotting within 6 min when incubated with whole blood, regardless whether autotransplantation, allotransplantation, or xenotransplantation was modeled. Data are mean ± SEM of at least four independent experiments. *p < 0.01. (B) The release of C-peptide, as a measure of β-cell damage, was significantly higher in xenogeneic (pig islets–human blood) than in allogeneic (human islets–human blood) combinations. *p < 0.05. Pig islets in culture medium or exposed to autologous blood released C-peptide in higher concentrations than human islets. The difference is likely the effect of the fragility of porcine islets and a different species-specific C-peptide kinetics of degradation.
C-Peptide Release as an Indicator of β-Cell Damage
Figure 1B shows the release of C-peptide during the 60-min in vitro experiments, comparing allogeneic versus xenogeneic islet-to-blood combinations. Exposure of pig islets to human blood caused a significant increase in release of pig C-peptide to 876 + 156 ng/ml (linear regression p < 0.0001, r2 = 0.48). In contrast, an increase of C-peptide from human islets was not observed (linear regression p > 0.05, r2 = 0.05). The release of C-peptide by pig islets was significantly greater than that by human islets, as evidenced by a difference in slope (p < 0.001) and higher mean values at 15, 30, and 60 min (Fig. 1B). The addition of epinephrine failed to decrease the release of C-peptide (data not shown), further indicating that C-peptide release was due to a membrane leakage rather than to a physiological response. We also measured C-peptide release in autologous combinations (pig islets and pig blood, n = 4), which was 235 ± 45 ng/ml after 60 min, thus significantly lower compared to the xenogeneic combination (p = 0.02).
Binding of IgM and IgG Antibodies and Activation of Complement
Cryo-sections of islets embedded in blood clots were fixed after 60 min and analyzed for antibody binding by immunofluorescence. IgM and IgG antibody staining was observed on pig islets incubated with human blood (Fig. 2A, B). No difference in this respect was observed when wild-type pig islets were compared with GT-KO islets (not shown). In contrast, antibody binding was virtually absent on pig islets incubated with autologous pig blood (Fig. 2C, D) and human islets incubated with allogeneic human blood (Fig. 2E, F).
Binding of human IgM and IgG antibody to pig islets (xenogeneic) (A–B) and to human islets (allogeneic) (E–F). Binding of pig IgM and IgG antibodies to pig islets (autologous) (C–D). IgM (green, A, C, E), IgG (green, B, D, F), insulin (red), nucleus (DAPI/blue). Yellow indicates colocalization of insulin and IgM/IgG.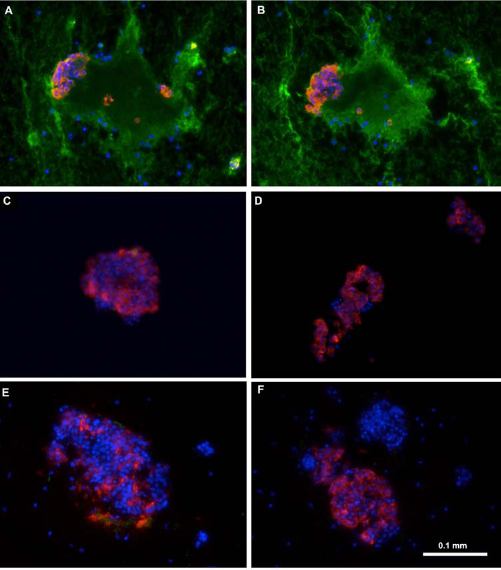
Complement activation, possibly subsequent to antibody binding, was assessed by measuring the soluble complement fragments iC3b, C4d, and Bb as markers of activated common, classical, and alternative complement pathways, respectively. Figure 3 compares the increments of iC3b and Bb in xenogeneic (pig islets–human blood) and allogeneic (human islets–human blood) experiments. Human islets did not significantly induce iC3b (linear regression p = 0.81, r2 = 0.02). Pig islets induced higher levels of iC3b at all time points; significance was reached for the 5- and 30-min time points (Fig. 3A). However, the slope of its regression line did not reach significance (linear regression p = 0.144, r2 = 0.56), likely due to variability between various samples. Human islets caused a low-grade increase in Bb over time (linear regression p = 0.018, r2 = 0.96). The increase caused by pig islets was also significant (linear regression p = 0.022, r2 = 0.96) and significantly greater than the one caused by human islets (p = 0.019). Mean levels differed at 30 and 60 min (Fig. 3B). No changes in C4d levels (fluid phase) were observed under any of the experimental conditions, and immunofluorescent staining for C4d (solid phase) remained negative (data not shown).
Supernatant levels of complement iC3b (A) and Bb (B) induced by pig islets (xenogeneic, n = 8) and human islets (allogeneic, n = 5). The baseline iC3b level measured in control blood was subtracted at each time point, so data represent true islet-induced complement activation. *p < 0.05.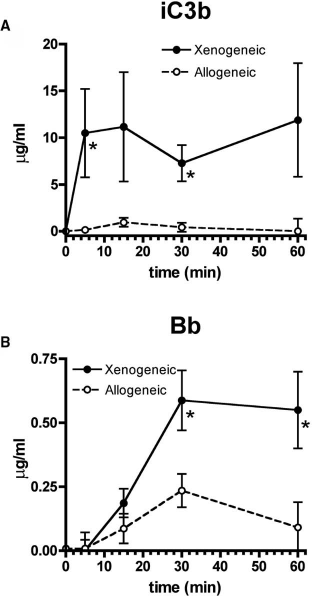
C5b-9, the membrane attack complex (MAC) that causes cell lysis, was more abundant on pig islets exposed to human blood than on human islets exposed to human blood (Fig. 4A, 1–2). Whereas the addition of LMW-DS attenuated but did not prevent C5b-9 deposition (Fig. 4B, 1), C5b-9 was not found after NAC, compstatin, or anti-IgM treatment (Fig. 4B, 2–4) nor on hCD46 transgenic islets (Fig. 4B, 5).
(A) Greater immunofluorescent C5b-9 (membrane attack complex) positivity was detected on pig islets exposed to human blood (1) when compared to human islets in contact with human blood (2). C5b-9 (red), insulin (green), nucleus (DAPI/blue). (B) C5b-9 staining on pig islets after treatment with: low molecular weight dextran sulfate (LMW-DS; 1), nacystelyn (NAC; 2), compstatin (3), and anti-IgM (4). Human cluster of differentiation 46 (hCD46) transgenic pig islet (5). C5b-9 (red), insulin (green), nucleus (DAPI/blue).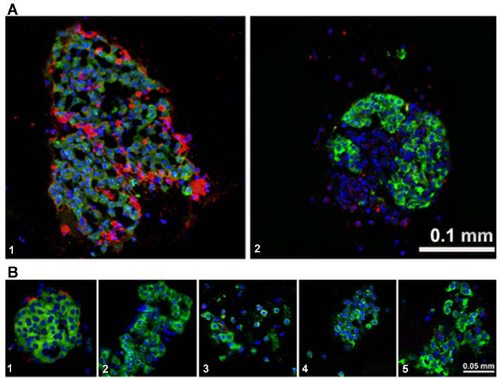
Effects of Modulating Treatments in Xenogeneic Blood–Islet Combinations
As Figure 5A–B shows, all therapeutic modalities, except the transgenic expression of hCD46 on the islets, significantly reduced iC3b and Bb levels. Lack of, or partial inhibition by, hCD46 could be related to its complement regulatory effect that occurs at a later stage in the complement cascade. Porcine C-peptide released after exposure of pig islets to blood (Fig. 5C) was significantly reduced by addition of compstatin and anti-IgM, but not significantly altered by LMW-DS, NAC, or hCD46 expression. The anticoagulant effect, in itself, by LMW-DS, therefore, did not prevent islet damage.
Levels of fluid phase (i.e., in supernatant) complement iC3b (A) and Bb (B), induced by pig islets exposed to blood for 30 min. Untreated versus LMW-DS, NAC, compstatin, anti-IgM, and hCD46 expression, respectively. All the therapeutic models, except hCD46 expression, significantly reduced complement activation. (C) C-peptide release after 30 min contact of pig islets with human blood. Compstatin and anti-IgM efficiently prevented C-peptide release, indicating a reduction in cell damage. *p > 0.06, †p < 0.05.
Effect of Exposure of Pig Islets to Human Plasma
Evidence that complement and antibody modulation resulted in reduced islet cell leakage led us to focus on humoral blood components and their possible involvement in early islet loss. Figure 6A shows that C-peptide concentrations, measured in the supernatant of pig islets exposed to human plasma, were comparable to those after exposure to whole blood. The lack of difference between whole blood and plasma suggests that early damage occurred as effects of humoral factors, which heat inactivation efficiently modulated (Fig. 6A).
(A) C-peptide release after contact of pig islets with human blood, plasma, or heat-inactivated plasma (H). Exposure of pig islets to human blood was compared to human plasma and human heat-inactivated plasma. The difference between plasma and heat-inactivated plasma was statistically significant. *p < 0.05. (B) Percentage of propidium iodide (PI)-positive (i.e., dead) islet cells after exposure to plasma or heat-inactivated plasma (H). Pig islets exposed to autologous plasma were used as control. Control versus plasma. *p < 0.05.
To further assess the extent of the damage, we quantified the number of PI-positive (i.e., dead) cells, comparing experimental (islets and plasma, islets and heat-inactivated plasma) with control conditions (islets and autologous plasma). Data showed (Fig. 6B) a statistically significant increase in PI-positive cells in plasma-exposed islets, while heat inactivation allowed for some protection (though statistically nonsignificant).
As Figure 7A shows, pig islets exposed to xenogeneic plasma had elevated basal mitochondrial respiration as well as respiration in the presence of oligomycin, when compared to autologous plasma, indicating increased leakage through the mitochondrial membrane. Increased membrane leakage translates into less production of ATP for cellular energetic needs. Exposure to xenogeneic plasma also caused a striking effect on the maximal respiratory activity of islet mitochondria by decreasing it twofold. All these observations are symptomatic of mitochondrial machinery dysfunction and are consistent with cell injury and islet apoptosis.
(A) Changes in oxygen consumption rates of pig islets exposed to xenogeneic plasma expressed as percentage of that of pig islets exposed to autologous plasma (set at 100%). At 5 mM glucose (Gluc5) and after exposure to oligomycin (Oligo), oxygen consumption was increased, indicating increased leakage through the mitochondrial membrane, consistent with cellular injury. The strongly reduced maximum respiration (Max Respiration) confirms this observation. (B) Ratios of respiratory parameters for plasma-treated islets (% of control). Islets exposed to xenogeneic plasma demonstrated a smaller response to high glucose as judged by the ratio of respiration in the presence of 20 mM glucose to basal respiration (G20/G5). They also had a smaller G20/Oligo ratio and a dramatic decrease in respiratory control (Max respiration/Oligo). These observations indicate impairment of insulin release and mitochondrial functions in these cells. *p < 0.05 and ***p < 0.001 compared to control.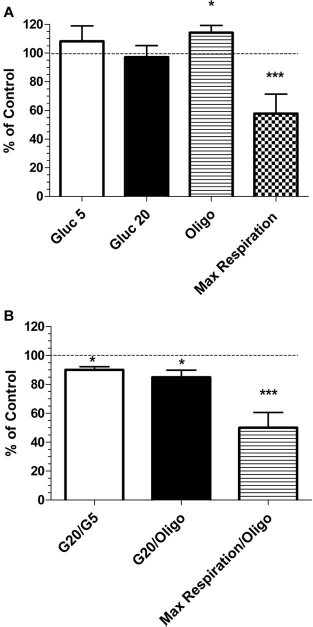
Plasma exposure was also associated with deposition of C5b-9 on islets (C5b-9 positivity), as well as IgM binding (Fig. 8A and B). Deposition of C5b-9 and IgM was reduced if the plasma was heat-inactivated (Fig. 8C and D).
C5b-9 and IgM deposition after exposure of pig islets to human plasma (A–B) and to heat-inactivated plasma (C–D). C5b-9 (red A and C), IgM (red B and D), insulin (green), nucleus (DAPI/blue).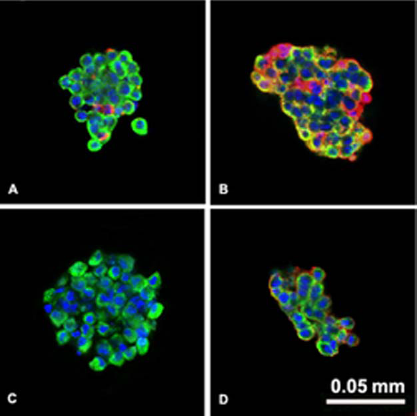
Discussion
The present study was aimed at characterizing the pathophysiological events triggered during direct exposure of pancreatic islets to blood, which occurs after islet Tx into the portal vein. Incubating pig and human islets with human blood proved a valuable model to investigate the events, as previously shown in a similar model by Dwyer et al. (16). A nonphysiological C-peptide release was observed in vitro, indicating acute β-cell damage, confirming similar in vivo reports after islet auto-, allo-, and xeno-Tx (34,45). The release of C-peptide from pig islets exposed to human blood was significantly greater than from equal numbers of human islets, indicating a greater lysis of the islets, which suggests that species incompatibility plays a pivotal role. Pig islets exposed to autologous blood released some C-peptide, which was higher than the amount released by human islets exposed to allogeneic blood. One possible explanation may be found in the fragility of porcine islets compared to human islets or in different kinetics of degradation of specie-specific C-peptides. We did not investigate this, as for our study, the effect of human blood/serum on pig islets was clinically more relevant.
Despite this difference in C-peptide release, activation of blood coagulation occurred equally rapidly in allogeneic and xenogeneic combinations. Even when autologous combinations were modeled, clotting occurred equally rapidly. However, anticoagulation did not prevent islet leakage, which correlates with previous reports that anticoagulation alone is insufficient to inhibit IBMIR and early islet loss (1,10). Nevertheless, anticoagulation to prevent portal vein thrombosis during islet intraportal infusion is certainly necessary.
Our next objective was to better understand the role of complement, which has previously been demonstrated to play a central role in IBMIR (4). In this in vitro model, levels of iC3b, an indicator of the early terminal complement pathway, were significantly increased when pig islets were exposed to whole human blood. The complement cascades led to the formation of the cytolytic product, the membrane attack complex (MAC or C5b-9). Pig islets exposed to human blood stained clearly positive for C5b-9. In contrast, human islets did not cause a significant increase of iC3b, and concomitantly, the extent of C5b-9 immunostaining was less.
Activation of the complement system can occur through several pathways, of which the classical and alternative pathways may be the most important (32). Eventually, both pathways lead to high iC3b and MAC formation. In xenogeneic combinations (i.e., pig islets with human blood), we evidenced involvement of both activation pathways. Pig islets exposed to human blood were positively immunostained for both IgM and IgG, indicative of the classical pathway. The use of GT-KO pigs (in association with the human CD46 transgenes) did not show any protective effect, suggesting that antibody binding to non-Gal antigens would likely be a potent complement activator. This correlates with the observation that natural anti-Gal antibodies do not appear to be detrimental to survival of adult pig islets (3,5,11,24) due to the low expression of Gal on these islets (14,41). Using a large particle flowcytometric technique, others also reported deposition of IgM and IgG antibodies on human and pig islets in in vitro models of IBMIR (22,44).
Activation of the alternative pathway depends on the subtle balance between spontaneously deposited low levels of C3b and exposure of cell surface complement regulatory molecules (33). When regulation of activation fails (as can be postulated when human complement binds to the pig cell surface due to incompatibilities between human complement and pig complement regulatory molecules), deposited C3b binds factor B, producing Bb. With the use of pig islets expressing hCD46, an attempt was made to inhibit the complement cascade. CD46 is a complement regulatory protein that is able to protect the host cell against complement injury by modulating proteolytic cleavage of C4b and C3b (2). Its features were maintained when transgenically expressed on pig cells (31). Human CD46 acts to restrict complement activation mediated by the alternative pathway on the cell surface rather than in the fluid phase (2). Indeed, in the current experiments, hCD46 expression on pig islets did not reduce fluid phase levels of Bb but appeared to reduce C5b-9 deposition on the islet cell surface. However, no clear protective effect regarding C-peptide release was observed.
In addition to direct membrane damage of β-cells, the inflammatory response is likely to induce a significant degree of apoptosis (19,27). Although this could be investigated in our in vitro model, it would be difficult to monitor in vivo after intraportal islet Tx. In contrast, C-peptide levels can be of immediate use after islet Tx as a measure of islet damage.
Our in vitro model was also intended to enable testing compounds that could exert a protective effect on the islets. Blockade of coagulation and complement by LMW-DS, NAC, and hCD46 were insufficient to fully prevent the release of C-peptide by pig islets exposed to human blood. Only compstatin and neutralizing anti-IgM antibodies modulated complement activation and prevented islet damage.
The need for efficient complement inhibition (e.g., with compstatin) and prevention of IgM binding to achieve islet protection indicated a relevant role for humoral factors. The experiments using human plasma further stressed this role. Xenogeneic human plasma triggered the same porcine C-peptide release as whole blood. Furthermore, plasma was sufficient to induce impairment of cell respiration and islet cell death. Mitochondrial dysfunction is likely among the mechanisms of cell death as a result of cell–plasma interaction. The effect of plasma could be efficiently prevented by heat inactivation. These observations strongly suggest that, in the pig-to-primate model, antibody binding and activation of the classical complement cascade may be playing a much more important role in early islet loss than previously considered.
Several strategies to prevent early islet loss are currently being investigated. Some aim at altering the interface between the islet surface and host environment, that is, by covering the islets with a heparin, endothelial cells, or polyethylene glycol (PEG) coating (10,28,43) or by alginate encapsulation (15). This might provide a barrier for antibody binding and complement activation. Administration of thrombomodulin prior to islet transplantation showed efficacy in modulating IBMIR (13). Pigs with multiple genetic modifications, including expression of complement regulatory and/or “antithrombotic” transgenes, as an alternative islet source may prove beneficial in reducing islet cell loss from IBMIR (16,38,46). Compstatin may soon be a clinically applicable complement inhibitor (39). If the important role of xenoreactive antibodies is confirmed in further (in vivo) experiments, pre-Tx antibody-neutralizing therapies (e.g., immunoadsorption, plasmapheresis, IVIg) may be investigated in nonhuman primate models of islet xeno-Tx. Finally, choosing a nonintravascular site, different from the portal vein, for transplantation of both allogeneic and xenogeneic islets may seem warranted to prevent IBMIR. Subcutaneous or intramuscular transplantation guarantees maximum patient safety and has become clinically applicable (reviewed in 47). However, gastrointestinal sites, such as the omentum, offer drainage of produced insulin into the portal vein for direct utilization in the liver (47). Our group has shown that IBMIR can be prevented in a pig allo-Tx model if islets are transplanted into the gastric submucosal space, a site where direct contact with the blood stream is avoided, but with a rich arterial blood supply for delivery of oxygen and nutrients (17).
Nonetheless, our data point to plasmatic factors as central players above and beyond procoagulative events and blood cellular interplay. Humoral immunity should be efficiently downregulated to prevent early islet loss and to make islet allo- and, particularly, xeno-Tx safe and successful forms of therapy.
Footnotes
Acknowledgments
The authors would like to thank Ms. Candy Knoll, Ms. Alexis Styce, and Mr. Bob Lakomy for excellent laboratory assistance and Drs. Hidetaka Hara, Chih Che Lin, Mohamed Ezzelarab, Yih-Jyh Lin, Eefje Dons, and Ms. Cassandra Long for help in performing pig donor pancreatectomies. This work was in part supported by Department of Defense grant W81XWH-06-1-0317 and JDRF grant 6-2005-1180. Sponsored research agreements between Revivicor, Inc., and the University of Pittsburgh exist. D.A. is CEO of Revivicor, Inc., and owns stock in the company. The authors declare no other conflict of interest.