
Abstract
Nonhematopoietic cord blood cells and mesenchymal cells of umbilical cord Wharton's jelly have been shown to be able to differentiate into various cell types. Thus, as they are readily available and do not raise any ethical issues, these cells are considered to be a potential source of material that can be used in regenerative medicine. In our previous study, we tested the potential of whole mononucleated fraction of human umbilical cord blood cells and showed that they are able to participate in the regeneration of injured mouse skeletal muscle. In the current study, we focused at the umbilical cord mesenchymal stromal cells isolated from Wharton's jelly. We documented that limited fraction of these cells express markers of pluripotent and myogenic cells. Moreover, they are able to undergo myogenic differentiation in vitro, as proved by coculture with C2C12 myoblasts. They also colonize injured skeletal muscle and, with low frequency, participate in the formation of new muscle fibers. Pretreatment of Wharton's jelly mesenchymal stromal cells with SDF-1 has no impact on their incorporation into regenerating muscle fibers but significantly increased muscle mass. As a result, transplantation of mesenchymal stromal cells enhances the skeletal muscle regeneration.
Introduction
Regeneration of mammalian skeletal muscle is the process leading to the reconstruction of muscle damaged as a result of exercise, mechanical injury, or degenerative diseases, such as muscular dystrophies. This process relies on the presence of “reserve” cells, also described as satellite cells or muscle precursor cells (56), which quiescently reside between sarcolemma and surrounding muscle fiber basal lamina. In response to muscle injury, satellite cells undergo activation, start to proliferate, differentiate, and fuse to reconstruct functional fibers. Each round of injury-induced satellite cell activation and differentiation is accompanied by their self-renewal and at least partial reconstitution of their population. Nevertheless, even in healthy organisms, the number of these cells gradually decreases during the lifetime. Importantly, in patients suffering from dystrophies (e.g., Duchenne's muscular dystrophy, Becker's muscular dystrophy), skeletal muscles undergo ongoing, multiple rounds of degeneration and reconstruction that, in consequence, lead to the exhaustion of satellite cell population and cause the failure in muscle regeneration.
Replacement of missing satellite cells became one of the goals of muscular dystrophy therapies. The cells of the “first choice” were myoblasts originating from satellite cells isolated from healthy donors (e.g., 36,41). However, it soon became clear that the ability of these cells to participate in muscle regeneration is impaired by their necrosis, apoptosis, and anoïkis (9,78), lack of ability to proliferate and migrate within the tissue of the recipient (e.g., 30,70), or host immune response (e.g., 30,71). Therapeutic potential of myoblasts was shown to be improved by certain experimental interventions. Thus, counteracting cell death (e.g., 10,28), enhancing cellular migration (e.g., 42,61,84), or impacting at immune system (e.g., 39,79,85) improved the ability of transplanted myoblasts to participate in muscle regeneration. Other approaches, such as use of scaffolds or gels (8,35) or manipulating the selected signaling pathways (e.g., 7,29), were also applied to prolong survival of transplanted satellite cells. Importantly, better results were achieved when freshly isolated satellite cells or muscle fibers with associated satellite cells, rather than myoblasts, were transplanted (e.g., 38,60,73). Other subpopulations of cells residing within the muscle were also shown to be able to undergo myogenic differentiation in vitro and participate in and improve muscle regeneration. Among them were mesangioblasts (62,76), pericytes (25), recently described muscle-resident interstitial cells that do not express paired box protein 7 (Pax7) but synthesize cell stress mediator PW1 [also known as paternally expressed gene 3 (Peg3)] (59), muscle side population cells (68), or so-called muscle-derived stem cells (MDSCs) (52). Moreover, even within the satellite cell population, only the limited proportion of cells was shown to be characterized by enhanced myogenic potential (72,82). In addition, other sources of “replacement” cells have been intensively tested. Among them are pluripotent embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) that were shown to be able to differentiate in vivo and also in vitro in virtually all known tissue types. However, their transplantation might cause the danger of their uncontrolled differentiation and teratoma development within the recipient tissue and thus creates the urgent need of the improvement of differentiation techniques. Despite that, the methods of iPSCs production undergo constant refinement; the most effective ones still involve infection of somatic cells with potentially dangerous retroviruses. Human ESCs, on the other hand, due to the methods of their generation, raise considerable ethical issues. As far as their application in the therapy of damaged muscle is concerned, the myogenic potential of pluripotent stem cells is limited, and generation of myoblasts requires additional procedures, such as purification of mesodermal progenitor cells (6,74), using of chemical agents (80), or overexpressing various factors such as Pax3 (20), or myogenic differentiation 1 (MyoD) (23). Among other promising sources of the cells that could substitute for satellite cells and engraft in regenerating muscle are stem cells isolated from human umbilical cord blood (HUCB) or mesenchymal stromal cells present in umbilical cord matrix, the so-called Wharton's jelly.
HUCB contains stem cells such as multipotent mesenchymal stem cells and hematopoietic stem and precursor cells. As a consequence of their stemness, they can reconstitute not only hematopoietic system (11) but also possess endothelial (91), osteogenic (46), or neurogenic potential (16). In our own study, we showed that HUCB cells are also able to participate in skeletal muscle regeneration (13). Another type of stem cells that can be isolated from the umbilical cord are cells residing within the Wharton's jelly, which is primitive embryonic type connective tissue surrounding umbilical cord blood vessels. Mesenchymal stromal cells isolated from Wharton's jelly were shown to be able to differentiate in vitro and in vivo into several cell lines, including hematopoietic cells (54), endothelial cells (90), neuronal cells (58), osteoblasts, chondroblasts, and adipoblasts (44). Using chromatinmodifying drugs, such as 5-azacitydine, can be induced to differentiate into myoblasts (18); thus, they can also be considered as a source of cells that could support skeletal muscle regeneration.
The present experiments were undertaken to test myogenic potential of umbilical cord-derived mesenchymal stromal cells (UC-MSCs), which is isolated from Wharton's jelly. First, we characterized the molecular status of these cells, that is, determined the expression of crucial pluripotency and myogenic markers as well as muscle-specific adhesion proteins. Next, we tested the hypothesis that UC-MSCs can undergo myogenic differentiation both in vitro and in vivo. Since existing data documents that SDF-1 (stromal-derived factor-1) treatment of stem cells, prior to their transplantation, increases their efficiency to colonize the injured tissue (33,86), we tested the hypothesis that stimulation of C-X-C chemokine receptor type 4 (CXCR4)/SDF-1 axis could be beneficial for both UC-MSC homing and muscle regeneration.
Materials and Methods
Human umbilical cord samples were processed according to the procedures approved by the institutional review board of the Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, with the donor consent. All animal studies presented were approved by Local Ethics Committee No. 4 in Warsaw, Poland.
Isolation, Culture, and Characterization of Umbilical Cord Mesenchymal Stromal Cells (UC-MSCs) Isolated From Wharton's Jelly
The term umbilical cords were collected during caesarean section surgical procedure after newborn's delivery. Sterile cords were transported at room temperature in containers filled with PBS supplemented with penicillin/streptomycin (Invitrogen Ltd., Paisley, UK). Cord sample, approximately 5 cm long, was first cut longitudinally, and blood vessels were removed. Resulting tissue was dissected and cut into pieces of 1–3 mm diameter, washed in high-glucose Dulbecco's modified Eagle's media (DMEM; Invitrogen Ltd., Paisley, UK), suspended in high-glucose DMEM supplemented with 20% of fetal bovine serum (FBS), and cultured in 37°C, 5% CO2, in flat-bottomed 25-cm2 culture flasks (Thermo Fisher Scientific, Nunc GmbH & Co. KG, Langenselbold, Germany). Original 5-cm cord sample was sufficient for starting cultures in three 25-cm2 flasks. After 5–7 days of culture, plastic-adherent cells were expanding around the Wharton's jelly fragments. Next, fragments were eliminated, and UC-MSCs were cultured in high-glucose DMEM supplemented with 20% FBS in 37°C, 5% CO2, passaged five to six times, and then harvested for mRNA extraction, coculture experiments, immunolocalization of selected epitopes, or transplantation. UC-MSCs potential to differentiate in vitro was analyzed. Cells were in vitro cultured in Stem Cell Technologies media, and then cells undergoing adipogenesis were visualized using Oil Red, chondrogenesis–Masson's Trichrome, and osteogenesis–Alizarin Red. All experiments were performed using the UC-MSCs originating from at least three umbilical cord collections from different donors.
Coculture of UC-MSCs with C2C12 Myoblasts
Murine C2C12 myoblasts (Sigma-Aldrich, St. Louis, MO, USA) were plated in six-well plates (2.5 × 103/cm2) and cultured in proliferation-inducing medium, that is, high-glucose DMEM supplemented with 10% of FBS, in 37°C, 5% CO2. Once C2C12 myoblasts reached confluence (more than 80%), UC-MSCs were added into the culture (1 × 103/cm2). At the same time, the proliferation- inducing medium was exchanged with differentiation- inducing one (32), that is, high-glucose DMEM supplemented with 10% FBS and 10% horse serum (HS, Invitrogen Ltd., Paisley, UK), and cells were cultured in 37°C, 5% CO2. After 4–5 days post UC-MSCs addition, the culture was terminated and cells were harvested for immunolocalization of selected epitopes. Control experiments involved simultaneous culture of UC-MSCs and C2C12 myoblasts alone, under the same experimental conditions as described above. Importantly, UC-MSCs did not spontaneously differentiate during a 14-day culture.
SDF-1 Treatment of UC-MSCs
UC-MSCs were cultured in high-glucose DMEM supplemented with 20% FBS. Once UC-MSCs reached confluence (more than 80%), the medium was supplemented with SDF-1 (0.2 μg/ml, Sigma-Aldrich). After 12 h of incubation, the cells were harvested for mRNA extraction or transplantation experiments.
Animals and Surgical Procedures
NOD.CB17-Prkdcscid/J (described as NOD/SCID throughout the text, i.e., non-obese diabetic severe combined immunodeficient) mice, purchased from Charles River Laboratories (France), were anesthetized with pentobarbital sodium salt (Sigma-Aldrich) in physiological saline by an intraperitoneal injection (30 mg/kg of body mass). Next, gastrocnemius muscle was injected with 25 ml of 10 μM cardiotoxin (CTX, Sigma-Aldrich) in physiological saline. Three days after CTX injection, 1 × 106 of UC-MSCs (untreated or SDF-1-treated) in 10 μl of physiological saline were transplanted into the CTX-injured muscle. Control muscles were either intact (n = 15) or CTX-injected only (n = 37). Regenerating muscles, isolated at days 7 (n = 29) and 14 (n = 30) after the procedure, were weighted and either subjected to the procedure of cell isolation or frozen for further RNA isolation or cryosectioning and immunolocalization analyses.
RT-PCR
Total RNA from the cells was isolated using High Pure RNA Isolation Kit (Roche Diagnostics GmbH, Mannheim, Germany) according to manufacturer's protocol. Total RNA from the muscles was isolated using RNeasy Midi Kit (Qiagen GmbH, Hamburg, Germany) according to manufacturer's protocol. The first-strand cDNA was synthesized and then amplified with Titan One Tube RT-PCR Kit (Roche Diagnostics GmbH, Mannheim, Germany) using 0.1 μg of total RNA as a template, with appropriate set of primers, specific for human sequences, under previously established conditions. We analyzed expression of octamer binding transcription factor 4 (Oct-4) (37), Nanog (50), Pax7 (81), MyoD (81), myogenic factor 5 (Myf5) (22), myogenin (22), integrin α3 (CD49c) (55), integrin β1 (CD29) (55), SDF-1 (1,5), CXCR4 (5,53), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (93) using the primers listed therein, which we sequenced to prove they also recognized the cDNA of the relevant species. Primers for the detection of M-cadherin were of our own design: forward 5′-TGACATTGCCAACTTCATCAG, reverse 5′-GATGAGAGCTGTGTCGTAGGG. Obtained products were separated through 1.5% Agarose LE gels (Roche Diagnostics GmbH, Mannheim, Germany) containing ethidium bromide. Optical densities of bands were analyzed using Gel Doc 2000 (Bio-Rad, Hercules, CA, USA) equipped with Quantity One software (Bio-Rad, Hercules, CA, USA). Levels of analyzed products were related to GAPDH product. Each analysis was performed at least three times, using mRNA isolated either from three different batches of in vitro cultured UC-MSCs or isolated from muscles collected during three independent regeneration experiments from at least three animals. All analyses were accompanied by positive controls in that transcripts were clearly detected by RT-PCR.
Histological Analysis
Muscles were dissected, frozen in isopentane cooled with liquid nitrogen, and stored at −80°C. Next, 10-μm-thick sections were obtained with cryomicrotome (Microm HM 505N, Thermo Fisher Scientific, Microm Inter national GmbH, Langenselbold, Germany). Sections were fixed with 3% paraformaldehyde (PFA, Sigma-Aldrich) in PBS, for 10 min, stained with Harris hematoxilin (Merck KGaA, Darmstadt, Germany) and eosin Y (Merck KGaA), analyzed using Eclipse TE 200 microscope (Nikon Instruments, Inc., Japan) equipped with NIS Elements F 2.30 (Nikon Instruments, Inc.) software. At least three muscles collected during three independent experiments were analyzed. In order to establish the proportion of immature and mature myofibers within the regenerating muscles, we counted the fibers within the surface of approximately 1 mm2. For each muscle, we analyzed three independent samples. At the 7th day of regeneration, we analyzed 1,413 (control muscles), 979 (UC-MSCs transplanted muscles), and 1,126 (SDF-1-treated UC-MSCs trans planted muscles) myofibers. At the 14th day of regeneration, we analyzed 1,118 (control muscles), 1,037 (UC-MSCs transplanted muscles), and 925 (SDF-1-treated UC-MSCs trans planted muscles) myofibers.
Immunolocalization
UC-MSCs and/or C2C12 cells in vitro cultured on sterile cover glasses in six-well plates or muscle sections were fixed with 3% PFA in PBS for 10 min and processed for immunolocalization of selected epitopes according to standard, previously described procedure (13,14). Chromatin was visualized either with DRAQ5 (Biostatus Ltd., Leicestershire, UK) or chromomycin A3 (Sigma-Aldrich). Samples were analyzed with confocal microscope Axiovert 100M (Carl Zeiss, Inc., Jena, Germany) equipped with LSM 510 software. Each immunodetection was performed at least three times using material collected during three independent experiments.
Cytometric Analysis of UC-MSCs—In Vitro Cultured or Isolated From Regenerating Muscle
UC-MSCs cultured in flat-bottomed 75 cm2 culture flasks were treated with 0.05% trypsin–EDTA (Invitrogen Ltd., Paisley, UK) for 5 min at 37°C. Next, isolated cells were washed three times in PBS, fixed in 3% PFA for 10 min, again washed in PBS, and processed for immunolocalization of selected epitopes according to standard, previously described procedure (13). Gastrocnemius muscle was dissected from leg of NOD/SCID mice, weighted, and fragmented with scissors. A single cell suspension was obtained by treatment of muscle fragments with 0.15% pronase (Sigma-Aldrich, St. Louis, MO, USA) in low-glucose DMEM containing 10% FBS (51). Cells were washed in PBS, fixed with 3% PFA in PBS for 10 min, and subjected to immunodetection according to previously described procedure (13). Flow cytometry analysis was performed using FACSCalibur instrument (Becton Dickinson, San Jose, CA, USA) equipped with 488-nm argon laser. Three parameters were acquired and stored forward scatter (FSC), side scatter (SSC), Alexa 488 (fluorescence 1, FL1). 1 × 1 0 5 events were collected for each sample. The background readings of either corresponding cells or corresponding muscle isolated from control animals were always subtracted. CellQuest 1.2 software (Becton Dickinson, San Jose, CA, USA) was used for the analysis.
Antibodies Used for Immunolocalization (IF) and Flow Cytometry (FACS)
Human cells were immunodetected using mouse anti-human nuclei (Millipore Chemicon, Billerica, MA, USA; MAB1281, IF, FACS) or phycoerythrin-labeled (PE) mouse anti-β2-microglobulin antibodies (Becton Dickinson, San Jose, CA, USA; 551337, IF, FACS). Laminin was immunolocalized with rabbit anti-laminin antibody (L9393; Sigma-Aldrich). General characterization of UC-MSCs included the analysis of the expression of selected surface cell markers: Lin1 (Becton Dickinson, 340546; Lineage cocktail including antibodies against CD3, CD14, CD16, CD19, CD20, CD56), CD29 (i.e., integrin β1; Becton Dickinson, 555443, FACS), CD34 (Becton Dickinson, 345802, FACS), CD45 (Becton Dickinson, 345809, FACS), CD73 (Becton Dickinson, 550257, FACS), CD105 (Caltag Laboratories, MHCD10504, FACS). More over, we used the following primary antibodies manufactured by Santa Cruz Biotechnology (USA): mouse anti-Oct-4 (sc-5279, FACS), rabbit anti-Nanog (sc-33759, FACS), mouse anti-Pax7 (sc-81648, FACS), rabbit anti-MyoD (sc-318, FACS), rabbit anti-Myogenin (sc-576, FACS), mouse anti-integrin α3 subunit (CD49c, sc-7019, FACS), rabbit anti-SDF-1 (sc-28876, FACS), followed by rabbit anti-mouse IgG Alexa Fluor 488, goat anti-rabbit IgG Alexa Fluor 488, and goat anti-rabbit IgG Alexa Fluor 594 (Molecular Probes, Invitrogen Ltd., Paisley, UK; A-11059, A-11008, and A-11037). All analyses were accompanied by the positive controls in that proteins investigated by us were clearly detected, and negative controls in that primary antibodies were omitted.
Statistical Analysis
Results were analyzed using SigmaStat 3.5. We have applied ANOVA and Student–Newman–Keuls post hoc test to evaluate the significance of the difference between each pair of means. A value of p < 0.05 was considered as statistically significant.
Results
Characteristics of UC-MSCs
Initial analyses of umbilical cord mesenchymal cells (UC-MSCs) (Fig. 1A) revealed that 80% of cells, after the first passage, and >95% of cells, after the third and fifth passages, could be characterized as cells expressing CD29, CD73, and CD105 and lacking CD34, CD45, and Lin1. These cells were also able to differentiate in vitro into adipogenic, chondrogenic, and osteogenic lineages (data not shown), thus confirming their previously described mesenchymal stromal cell characteristics (26,40).
Characteristics of human UC-MSCs. (A) Morphology of UC-MSCs (umbilical cord-mesenchymal stromal cells) in vitro cultured for 4 days. Scale bar: 200 μm. (B) Analysis of the presence of mRNAs encoding selected pluripotency markers, myogenic factors, adhesion molecules, stromal derived factor (SDF)-1, and its receptor C-X-C chemokine receptor 4 (CXCR4). (C–H) Proportion of cells expressing chosen markers determined by fluorescence activated cell sorting (FACS) analysis (representative graphs): (C) octamer-binding transcription factor 4 (Oct-4), (D) Nanog, (E) paired box protein 7 (Pax7), (F) mygenic differentiation 1 (MyoD), (G) myogenin, and (H) integrin α3.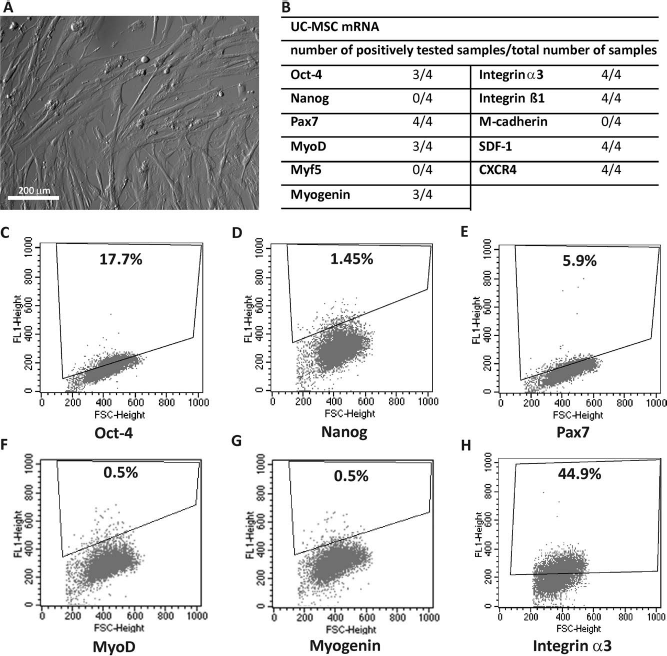
Next, we focused at the expression of factors necessary for retaining pluripotency and factors crucial for myogenic differentiation. To this end, we analyzed the expression of Oct-4 and Nanog transcription factors, characteristic for pluripotent stem cells, as well as factors characteristic for the specification and differentiation of myogenic precursor cells and myoblasts, such as Pax7, MyoD, Myf-5, and myogenin, as reviewed by Buckingham and Vincent (15). Moreover, we evaluated the presence of selected adhesion molecules that are known to be markers of mesenchymal cells and also has been shown to be involved in myoblast adhesion and fusion, that is, α3 and β1 integrin subunits (12,77) and myoblast-specific M-cadherin (27). Last but not least, we determined the expression of SDF-1 cytokine and its CXCR4 receptor that plays a crucial role in various stem cell migration and homing (48).
Oct-4 encoding mRNA was detectable only in three of four UC-MSC samples (Fig. 1B); however, Oct-4 protein was expressed only in small proportion of analyzed cells. FACS analysis allowed us to determine that barely 13.1% (±7.45%) of the cells expressed this protein (Fig. 1C). We failed to detect mRNA-encoding Nanog (Fig. 1B) and also to immunolocalize this protein in in vitro cultured UC-MSCs (not shown). However, by FACS we detected Nanog-positive cells in two of three analyzed samples, but in a very low number, which are 1.45% and 5.3%, respectively (Fig. 1D). Pax7 transcripts were present in all UC-MSCs samples studied (Fig. 1B), and Pax7-positive cells in two of three samples analyzed by FACS (5.9% and 5.2% of analyzed cells, respectively) (Fig. 1E). MyoD and myogenin mRNA were expressed in three of four samples (Fig. 1B), but only approximately 0.5% of cells analyzed by FACS express these factors (Fig. 1F, G). We were not able to confirm the presence of neither mRNA-encoding Myf-5 nor M-cadherin. But we detected α3 (Fig. 1B, H) and β1 integrin subunit expression (Fig. 1B, not shown). UC-MSCs expressed SDF-1 and CXCR4 receptors, both at mRNA (Fig. 1B) and protein level (not shown). In summary, only a small fraction of UC-MSCs expressed Oct-4 and Nanog and, thus, could be considered as potentially pluripotent, that is, as “true” stem cells. Moreover, basing on the fact that also a very small proportion of cells were Pax7-positive, even a smaller number expressed MyoD or myogenin, and that they did not express M-cadherin, we could not describe them as a cells readily representing “myogenic” potential.
In Vitro Differentiation of UC-MSCs
To verify our hypothesis that human UC-MSCs are able to differentiate into myoblasts, we cocultured these cells in the presence of differentiating mouse C2C12 myoblasts. Each experiment was accompanied by control cultures of UC-MSCs and C2C12 myoblasts. Control mesenchymal stromal cells remained undifferentiated, while C2C12 cells initiated myogenic differentiation. UC-MSCs cocultured with C2C12 myoblasts were fixed and analyzed at day 5 (proliferation), day 7 (early stages of cell fusion), and day 9 (formation of myotubes) after the beginning of coculture. Human cells were visualized by immunodetection either of human β2-microglobulin or human nuclear antigen, using antibodies that were proved to detect human antigens (13). Thus, only UC-MSCs and myotubes originated from them were labeled. At day 5 of coculture, UC-MSCs were randomly positioned among proliferating C2C12 myoblasts (Fig. 2A, B). At day 7, UC-MSCs started to align (Fig. 2A, B), and at day 9, myotubes containing nuclei labeled with antibody recognizing human nuclear antigen and expressing human β2-microglobulin were formed (Fig. 2A, B). However, the fact that nuclei-expressed human nuclear antigen did not necessarily mean that all of them were of human origin. It was previously shown that, in heterokaryons between mouse C2C12 cells and human fibroblasts, all nuclei, regardless of their origin, expressed human-specific antigen (e.g., 65). In our analyses, however, we also found myotubes containing only one nucleus labeled with antibody detecting human nuclei antigen among nonlabeled ones (Fig. 2A). Nevertheless, we decided that we are not able to reliably establish how many UC-MSCs and C2C12 myoblasts participated in the formation of each myotube. Thus, we quantified only the frequency of myotubes containing human nuclei. By doing that, we revealed that 15% (10 of 67) of them were hybrid ones, which allowed us to conclude that UC-MSCs are able to differentiate into myoblasts and to form myotubes under the conditions stimulating myogenic differentiation.
In vitro differentiation of UC-MSCs. UC-MSCs cocultured with C2C12 myoblasts were fixed at different stages of their differentiation, that is, during the period of their proliferation (top), fusion (center), and when the multinuclear myotubes were formed (bottom). Human cells were visualized by immunodetection of either human β-2-microglobulin (A) or human nuclear antigen (B). (A) Immunolocalization of human β-2-microglobulin (red), visualization of nuclei (blue), overlaid with transmitted light image. (B) Immunolocalization of human nuclear antigen (green), visualization of nuclei (blue), overlaid with transmitted light image (left column only). Arrows indicate the position of human nuclei within multinucleated myotube. Scale bar: 20 μm.
Influence of Cell Transplantation on Muscle Regeneration
In our study, cells were transplanted to cardiotoxin (CTX)-injured m. gastrocnemius of immunodeficient NOD/SCID mice that were shown to accept xenotransplants. This experimental design allowed us to easily trace human cells within mouse tissues using anti-human nuclei antibodies. In our previous study, we carefully followed regeneration of mechanically injured muscles of SCID mice and showed that this process did not differ from the muscle regeneration of BALB/c mice (13). In the preliminary experiments, we confirmed that the same remains true for NOD/SCID cardiotoxin injured skeletal muscle in that their regeneration is the same as that of BALB/c CTX-injured muscle. At the first stage of experiments, we compared the progress of regeneration of control, CTX-injured skeletal muscles, and CTX-injured skeletal muscles to which UC-MSCs were transplanted. Several lines of evidence and also our own studies showed that CXCR4–SDF-1 axis plays a crucial role in the migration and homing of various stem cells (13,48). Thus, we decided to pursue the possibility that homing of SDF-1-treated cells will be more efficient and will influence the regeneration processes. To test that notion, we performed the experiment in that in vitro cultured UC-MSCs were pretreated with SDF-1 before transplantation. Analyses of SDF-1 and CXCR4 expression in untreated and SDF-1-treated UC-MSCs showed that the level of mRNA encoding SDF-1 was comparable in both groups of cells. However, expression of CXCR4 increased significantly after the SDF-1 treatment, suggesting that the cells reacted to the presence of the cytokine (Fig. 3A). In each experiment, untreated and SDF-1-treated UC-MSCs were transplanted into m. gastrocnemius 3 days after CTX injection. Next, muscles were analyzed at days 7 and 14 after the cell transplantation.
Analysis of regenerating muscles implanted with UC-MSCs. (A) RT-PCR analysis of CXCR4 and SDF-1 levels in in vitro cultured control and SDF-1-treated UC-MSCs. Optical density of the products separated in agarose gels are presented as a percentage of the optical density (Odu) of glyceraldehyde 3-phosphate dehydroegenase (GAPDH) amplification product. (A) Comparison of weight of regenerating control muscles and muscles to which UC-MSCs and UC-MSCs treated with SDF-1 were implanted. Muscles were analyzed at days 7 and 14 of regeneration. Results were analyzed using ANOVA and Student–Newman–Keuls post hoc test to evaluate the significance of the difference between each pair of means. Upper indexes indicate statistical significance: 1p = 0.036; 2p < 0.001; 3p = 0.011. (C) Histology of m. gastrocnemius at days 7 (left) and 14 (right) of regeneration to which none of the cells were injected (control, top) and those injected with UC-MSCs (middle) and UC-MSCs treated with SDF-1 (bottom). Hematoxylin/eosin staining. Scale bar: 200 μm. (D) Histology of noninjured m. gastrocnemius. The muscle is composed of fast (light gray) and slow (dark gray) twitch myofibers. Hematoxylin/eosin staining. Scale bar: 200 μm. (E) Comparison of the proportions of immature (centrally positioned nuclei) and mature (peripherally positioned nuclei) myofibers within the regenerating muscles analyzed at days 7 and 14 of regeneration (C).
Analyses of muscle weight revealed that intact, noninjured m. gastrocnemius weighted 0.142 g (±0.014 g, n = 15). At day 7 after CTX injury, the weight of control, which is the injured and regenerating muscle, did not differ significantly neither from the weight of control muscles nor from regenerating ones to which UC-MSCs or SDF-1-treated UC-MSCs were transplanted (Fig. 3B, ANOVA). However, at day 14 after CTX injury, differences in the muscle mass become significant. Transplantation of all cell types caused increase in the weight of regenerating muscle, as compared to control, injured muscle that did not receive any cells (Fig. 3B, ANOVA and Student–Newman–Keuls post hoc test). Moreover, the muscles injected with untreated and SDF-1-treated UC-MSCs differed significantly from each other (Fig. 3B). Histological analyses of CTX-injured muscles to which none of the cells were implanted and muscles that received untreated or SDF-1-treated UC-MSCs revealed beneficial effect of cell transplantation on the regeneration process (Fig. 3C, D). At day 7 and also at day 14 of regeneration, less regenerating (i.e., centro-nucleated) myofibers were visible in control muscles, as compared with ones that received SDF-1-treated cells. We compared the number of immature (i.e., centro-nucleated) and mature (i.e., containing peripherally positioned nuclei) myofibers. These analyses showed that, in muscles that received SDF-1-treated UC-MSCs, the number of myofibers with peripherally positioned nuclei (i.e., mature ones) was lower than in control. Importantly, this effect was already noticeable at day 7 of regeneration (Fig. 3E). Nevertheless, even at day 14 of regeneration, none of the muscles exhibited the morphology of noninjured m. gastrocnemius (i.e., regular organization of fast and slow twitch myofibers with peripherally positioned nuclei) (Fig. 3C, D). Histological analyses were accompanied by the examination of the level of mRNAs encoding CXCR4 and SDF-1 that was performed on intact (i.e., control) and regenerating muscles that received untreated or SDF-1-treated UC-MSCs. These analyses showed that muscle injury resulted in the rise of the levels of both analyzed transcripts, at day 7 of regeneration. Observed increase was similar in control and in UC-MSC-injected muscles, regardless of SDF-1 treatment (Fig. 4). However, at day 14 after injury, control muscles expressed lower levels of CXCR4 mRNA than those that receive UC-MSCs. Thus, cell transplantation improved muscle regeneration, as judged by the significant increase in muscle weight, their histology, and expression of SDF-1 and its receptor.
CXCR4 and SDF-1 expression in intact and regenerating muscles. CXCR4 and SDF-1 expression levels were analyzed by RT-PCR in intact (noninjured), control (injured), UC-MSCs, and UC-MSCs treated with SDF-1-injected muscles. Analyses were performed at days 7 (7R) and 14 of regeneration (14R). Results were presented as a percentage of the optical density (Odu) of GAPDH amplification product.
In Vivo Differentiation of UC-MSCs
Analysis of the number of UC-MSCs residing within the regenerating muscle was performed using flow cytometry. It enabled us to determine the proportion of human cells among the cells that were mononuclear or contained only few nuclei, isolated from regenerating muscle. Such analysis revealed that approximately 30% of the cells, isolated from the muscle at day 7 of regeneration, were of human origin (Fig. 5A). Similar proportion was calculated after the analysis of the cell isolated from muscles to which SDF-1-treated UC-MSCs were injected (Fig. 5A). Thus, at day 7 of regeneration, the SDF-1 pretreatment did not influence the number of human cells sustained within the regenerating muscle.
UC-MSCs in regenerating muscles. (A) Representative graphs showing results of FACS analysis of cells isolated from control, UC-MSCs, and UC-MSCs treated with SDF-1-injected muscles at day 7 of regeneration. Proportion of positive cells among cells counted was indicated in each graph. Human nuclei were detected with antibody specifically recognizing human nuclear antigens. (B) Control regenerating muscle contains no human nuclei (left). Human nuclei present within muscles injected with UC-MSCs (middle) and UC-MSCs treated with SDF-1 (right) at days 7 (top) and 14 (middle and bottom) of regeneration. Human nuclei were randomly localized within the regenerating muscle (day 7 of regeneration, right, arrows), found within forming myofibers (day 7 of regeneration, middle, arrow; day 14 of regeneration bottom, arrows), or they were found within in the intimate vicinity of muscle fibers (days 7 and 14 of regeneration, inserts, middle and right). Red, human nuclear antigen; blue, nuclei; green, laminin. Scale bar: 50 μm; scale bar in inserts: 10 μm.
To test the ability of UC-MSCs to participate in the formation of the myofibers in vivo using antibody specifically recognizing human nuclei, we localized these cells within sections of regenerating muscles. At day 7 of regeneration, in majority of analyzed muscle sections, both untreated and SDF-1-treated UC-MSCs, produced a cell mass restricted to the close vicinity of newly formed myotubes (Fig. 5B). At day 14 of regeneration, we did not observe such accumulation neither of untreated or SDF-1-treated cells. Sporadically, we observed cells included into the regenerating myofibers. However, the frequency of such events was low, (i.e., 5.3%, 3 of 57) of analyzed myofibers contained human nuclei. At both time points, we detected UC-MSCs localized in the close vicinity of muscle fibers, in the positions resembling satellite cells (Fig. 5B). At days 7 and 14 of regeneration, 4.2% (9 of 215) and 3.9% (8/206) of human cells were found in such sites, respectively.
Discussion
Stem cells that can be considered as a suitable material for transplantation and replacement therapies not only have to be pluri- or multipotent, that is, able to differentiate into at least few cell types, but also should be easily maintained and propagated in vitro. Several lines of evidence documented that mesenchymal stromal cells, isolated from umbilical cord blood or mesenchymal stromal cells from Wharton's jelly, fulfill this requirement. In the current work, we tested the myogenic potential of mesenchymal stromal cells isolated from umbilical cord (UC-MSCs) and proved that these cells are able to undergo myogenic differentiation in vitro, but they only rarely participate in the reconstruction of damaged skeletal muscle. However, their presence significantly improved the regeneration process.
First of all, we tested UC-MSCs' pluripotent potential in analyzing the expression of pluripotency markers, such as Oct-4 and Nanog, that were reported to be expressed by ESCs, iPSCs (e.g., 89), very small embryonic-like cells (VSELs) (49), and also by some tissue-specific progenitors (e.g., 92). We showed that in each analyzed sample, Oct-4 and Nanog were synthesized only by small fraction of UC-MSCs. Thus, our data strongly suggested that only a small fraction of UC-MSCs might have characteristics and a potential of “model” stem cells. Similar conclusions came from the study on porcine umbilical cord matrix cells; among that, only a very small proportion expressed Oct-4 (17). The rarity of pluripotent cells can also be a reason why mesenchymal stromal cells isolated from Wharton's jelly fail to form embryoid bodies. These structures, containing cells of ecto-, endo-, and mesodermal characteristic, can be generated from ESCs or iPSCs. However, Wharton's jelly cells, cultured under the conditions stimulating the formation of embryoid bodies, form spheres that degenerate after a couple of days of culture (31). Lack of proper “pluripotent” characteristic does not, however, prevent differentiation of mesenchymal stromal cells into various cells types, including myoblasts, as we documented in the present study. Myogenic potential of UC-MSCs might be suggested by the fact that these cells express myogenic markers. It has to be noted, however, that only a limited population of these cells was Pax7, MyoD, or myogenin positive and that they did not express M-cadherin or Myf-5. We did detect expression of adhesion proteins, such as integrin α3 and β1 subunits, that are not unique for myogenic cells but, as it was shown by us and others, are crucial for myoblast fusion and formation of myotubes (12,77). Importantly, UC-MSCs express SDF-1 ligand and its receptor CXCR4, which plays a crucial role in homing of migrating stem cells and which we previously showed to participate in skeletal muscle regeneration (13).
Myogenic Differentiation
Transplantation of cells into injured and regenerating muscles were preceded by experiments in that UC-MSCs were cocultured with differentiating C2C12 mouse myoblasts. Such experimental system did not require chemical intervention, such as the use of chromatinmodifying drug 5-azacytidine (19) or dexametasone- and hydrocortisone-supplemented medium (34), which were shown to induce myogenic differentiation of mesenchymal cells isolated from HUCB or Wharton's jelly. By coculturing UC-MSCs with C2C12 myoblasts, we proved that these cells indeed exhibit myogenic potential and are able to form myotubes. As a result, we observed 15% of hybrid myotubes, which is formed with the participation of both UC-MSCs and C2C12 myoblasts. Similar experimental system was applied for CD34+ HUCB cells cultured with myoblasts of patients suffering from Duchenne's or Becker's muscular dystrophies (43). Such culture conditions also promoted fusion between CD34+ HUCB cells and dystrophic myoblasts. As a result, the myotubes in that dystrophin synthesis were formed. Moreover, CD105+ Wharton's jelly cells were shown to be able to undergo osteogenic, adipogenic, and also myogenic differentiation in vitro (18).
Our transplantation experiments showed that UC-MSCs implanted into regenerating muscle were able to survive in such environment. At day 7 of regeneration, approximately 30% of mononucleated cells isolated from skeletal muscles and analyzed by FACS were human cells. Moreover, histological analyses of such muscles revealed that UC-MSCs did not perturb the repair process. Their initial localization in the vicinity of newly formed myotubes suggested that they might be able to interact and participate in the regeneration. However, in the course of the regeneration, these cells dispersed within the muscle, and only some of them were found localized in the close vicinity of muscle fibers. Similar localization was reported for the human bone marrow mesenchymal stromal cells transplanted to the injured tibialis anterior muscle (24). Moreover, the so-called PICs (PW1-expressing interstitial cells), which were shown to participate in muscle regeneration, also occupy akin interstitial position (59).
Previously, SDF-1 was shown to improve the migration ability of transplanted cells (33) and to impact at the migration of circulating cells towards the site of the injury (67). Thus, we subjected UC-MSCs to this cytokine before their transplantation into the site of the injury. Importantly, SDF-1 treatment increased the level of CXCR4 expression in UC-MSCs. Since it was previously reported that CXCR4 positively influence the engraftment of muscle progenitor cells into reconstructed muscle fibers (68), we reasoned that its increased level will also be beneficial for UC-MSCs' participation in the regeneration. Unfortunately, we did not observe increased incidence of hybrid myotubes in muscles transplanted with SDF-1-treated UC-MSCs. Regardless of SDF-1 treatment, hybrid muscle fibers were found only sporadically. Since SDF-1 did not increase the incidence of hybrid myofibers originating from UC-MSCs, we reasoned that it might facilitate the colonization of the injured muscle by endogenous cells. This phenomenon can be explained by the ability of mesenchymal stromal cells to change the environment of the regenerating muscle, for example, via local increase of SDF-1 concentration. Moreover, SDF-1 pretreatment of UC-MSCs could stimulate positive feedback to produce even more SDF-1 and CXCR4 and, by local rise of the level of these factors, increase stem cell migration and homing, as reviewed in Kucia (48).
Other studies focusing at the potential of mesenchymal cell populations also document that such cells, however, with various success rate, can participate in skeletal muscle regeneration. For example, CD105+ cells from Wharton's jelly were detectable as long as for day 14 after the injection into the injured rat tibialis anterior muscle (18). Transplantation of human umbilical cord tissue into the caudal vain of sjl mice lead to the expression of dystrophin, as judged by RT-PCR and Western blot analyses, and also improvement in muscle strength, suggesting that these cells were able to participate in the formation of functional myofibers (88). Human umbilical cord blood cells were able to undergo myogenic differentiation in the environment of dystrophic muscles of mdx or sjl mice, which resulted in the generation of muscle fibers expressing dystrophin or dysferlin (47,64). CD34+ cord blood cells injected into ischemic mouse hind limb not only accelerated the muscle repair in vivo but also participated in the formation of new muscle fibers with the success rate of 5.6% (69), which is similar to that achieved by us. Human adipose-derived mesenchymal stromal cells injected into muscle of mdx mice were able to differentiate into skeletal muscle fibers and restore dystrophin expression (87). Interestingly, despite that in our study UC-MSCs cells did not form numerous myotubes containing human nuclei within the regenerating muscles, they significantly improved the regeneration, had beneficial effect on the muscle mass, and resulted in the increase of mature myofibers in regenerating muscles. Such increase of muscle mass was observed in other studies after myoblast (4), or bone marrow implantation (75). In the latter case, bone marrow transplantation reduced the myopathic phenotype of SMA (spinal muscular atrophy) mouse model. Similar to our study, examined cells also only rarely participated in the formation of new muscle fibers. Thus, the improvement of muscle regeneration was attributed to so called “biological activity” of bone marrow cells (75). Also in other studies, bone marrow mesenchymal stromal cells significantly enhanced regeneration of injured rette tibialis anterior muscle without participation in this process (63). Such “biological activity” relies at paracrine secretion of trophic factors, that is, growth factors, and cytokines that regulate migration, proliferation, and differentiation of endogenous cells, that is, satellite cells and also other muscle-associated cells, as reviewed by Ten Broek et al. (83). These factors of course can be produced not only by exogenous cells but also by endogenous ones, that is, satellite cells, myoblasts, and also inflammatory cells, fibroblasts, as well as other ones (reviewed in 66,83). Among factors that have been shown to be involved in myoblast differentiation and muscle healing are basic fibroblast growth factor (bFGF), insulin-like growth factor-1 (IGF-1), hepatic growth factor (HGF), interleukin 6 (IL-6), and other ones (e.g., 45,57). Importantly, these factors can influence muscle regeneration not only by acting on myoblast but also by promoting vascularization (e.g., 21). For example, injection of human adult multipotent progenitor cells (hMAPCs) into the ischemic murine muscle improved its regeneration not only by impacting at myoblast differentiation but also by exerting trophic arterio/angiogenic effect (2,3). Moreover, in vitro analyses revealed that hMAPCs were able to prevent myoblasts apoptosis (3).
Changes in muscle weight observed in our study could be, therefore, caused by modes of action other than the “myogenic” one, that is, other than fusion of UC-MSCs with newly formed myotubes. Such “trophic” mechanism might be beneficial in cases when regeneration enhancement is sought but cannot be considered beneficial for the patients suffering from such conditions as Duchenne's muscular dystrophy caused by the lack of functional dystrophin gene in all cells. In such cases, only the cells that can be easily propagated in vitro and with high efficiency contribute to new myofiber formation and then resurrect dystrophin synthesis should be sought. Cells tested by us indeed present myogenic potential, but more work needs to be done to optimize conditions of their myogenic differentiation in vivo.
In summary, we can state that under the influence of in vitro culture conditions UC-MSCs were able to undergo myogenic differentiation and fuse to form the myotubes. However, under the in vivo muscle microenvironment conditions, these cells are only sporadically included into newly reconstructed myotubes, but their presence had a beneficial effect on the injured muscle regeneration.
Footnotes
Acknowledgments
This research was supported by the Polish Ministry of Science and Higher Education grant N 301 405 133. We would like to thank Katarzyna Ilach for her technical assistance during cell culture experiments, Magdalena Ambrozek for help with cytometric analyses, Danuta Wasilewska for taking care of laboratory animals, and Maciej Krupa for help with confocal microscopy imaging. We also thank all members of the Department of Cytology for their constant support. Authors declare no conflicts of interest.