
Abstract
Wharton's jelly is a known stem cell source in humans. Because stem cells might provide a potential therapeutic role in canines, many stem cell sources are studied for isolation and characterization in the canine system. So far, there have been no reports identifying canine Wharton's jelly stem cells. In this study, we successfully isolated and characterized mesenchymal stem cells (MSCs) from canine Wharton's jelly. Canine Wharton's jelly-derived mesenchymal stem cells (cWJ-MSCs) that were grown in low-glucose DMEM medium have spindle-like shapes similar to human Wharton's jelly stem cells. We characterized the immunophenotypes of canine Wharton's jelly stem cells by FACS analysis and measured the cumulative population doubling level (CPDL). We investigated the differentiation of cWJ-MSCs with a trilineage differentiation assay to determine whether they were mesenchymal. Under various differentiation conditions, cWJ-MSCs presented chondrogenic, osteogenic, adipogenic, and neurogenic differentiation abilities in vitro. In conclusion, our results show that cWJ-MSCs might be a good source for stem cells. Furthermore, cWJ-MSCs might be useful as a cell therapy application for veterinary medicine.
Introduction
In humans, Wharton's jelly, called umbilical cord tissue, can be used as a source of mesenchymal stem cells (MSCs) (13,20,29). MSCs are capable of multilineage differentiation and self-renewal. MSCs, which are the most well-known stem cells, can be found in many tissues, including bone marrow, adipose tissue, the placenta, blood vessels, skeletal muscle, skin, and the brain (4,8,11,21,25,28,30). Although MSCs have been identified from multiple tissue sources, such as adipose tissue, umbilical cord blood, and bone marrow in the canine system (23,27,30), there have been no reports about MSCs from canine Wharton's jelly.
In stem cell biology, Wharton's jelly-derived MSCs have been suggested to be a promising source for stem cells to be used for regenerative medical applications in humans (24,26). Wharton's jelly-derived MSCs showed promising results in response to an animal model for a hepatic disease, both in vitro and in vivo (2,14).
Recently, some reports have shown that canine MSCs have therapeutic effects in studies of veterinary medicine regenerative disease (1,22). In the present study, we suggest that characterized canine Wharton's jelly-derived mesenchymal stem cells (cWJ-MSCs) represent a good model system for stem cell research in veterinary medicine. Furthermore, cWJ-MSCs could be used, therapeutically, in canine regenerative medicine studies. Also, we suggest that canine Wharton's jelly could represent a good source for MSCs that should be useful for dogs.
Materials and Methods
Animals
Healthy adult mixed-breed dogs (n = 4; 4.5 ± 0.4 kg) were used. Applicable institutional and governmental regulations concerning the ethical use of animals were followed during the course of this research. This investigation was performed in accordance with the Care and Use of Laboratory Animals guidelines of Seoul National University. In the cesarean section delivery, the dogs were premedicated with acepromazine maleate (0.1 mg/ kg; Sedaject, Samwoo medical, Yesan, Korea) and then thiopental sodium (15 mg/kg; Pentotal, Joongwei Pharmaceutical, Seoul, Korea) was injected intravenously to induce anesthesia. Isoflurane (AErrane, Baxter, Mississauga, ON, Canada) was used to maintain anesthesia. The procedure was performed under sterile conditions.
Cell Isolation and Culture
Cell isolation was performed as previously described with some modifications (24). All samples of canine Wharton's jelly were collected from cesarean section deliveries. Wharton's jelly is cord matrix, which could be easily distinguished from placenta by its macrostructure. The collected tissue was delivered into the sterile specimen cup and then washed with 0.9% normal saline to remove as much blood as possible. Blood vessels were mechanically removed with thumb forceps. After that, the canine Wharton's jelly tissue was finely minced, using a surgical blade, and resuspended in digestion solution [collagenase type I (2 mg/ml); Worthington Biochemical, Freehold, NJ]. The minced tissue was incubated at 37°C for approximately 3–4 h. After incubation, the suspension was washed with phosphate-buffered saline (PBS; Cellgro, USA) and centrifuged at 350 × g for 5 min. The pellet was resuspended in the basal culture medium, which is low-glucose Dulbecco's modified Eagle's medium (LG-DMEM; Gibco BRL, USA) with 10% fetal bovine serum (FBS; Gibco BRL, USA). The resuspended cells were seeded into T75 polystyrene cell culture flasks (Nunc, USA) with basal culture medium. The basal culture medium was replaced three times per week until the adherent cells reached 70–80% confluency.
Cumulative Population Doubling Level Analysis
The estimated growth efficiency and proliferation potential of cWJ-MSCs were determined by the total cumulative population doubling level (CPDL) using the formula CPDL = ln(Nf/Ni)ln2, where Ni is the initial seeding cell number, Nf is the final harvest cell number, and ln is the natural log. Cells (5 × 104) were plated in triplicate in a six-well culture plate (Nunc) and subcultured 5–7 days later. The final cell numbers were counted and 5 × 104 cells were replated. To yield the cumulated doubling level, the population doubling for each passage was calculated and then added to the population doubling levels of the previous passages.
Flow Cytometry
The cWJ-MSCs were stained for flow cytometry with specific antibodies following the protocol provided by the supplier (BD Biosciences, USA). In brief, cultured cWJ-MSCs were washed two to three times in PBS and harvested using 0.25% trypsin/EDTA. The cells were washed with PBS and divided into groups for antibody staining. Each aliquot contained approximately 1 × 106 cells. Mouse anti-human CD3, mouse anti-human CD11c, mouse anti-human CD28, mouse anti-human CD34, mouse anti-human CD38, mouse anti-human CD41a, mouse anti-human CD45, mouse anti-human CD62L, mouse anti-human CD90 (BD Biosciences), and mouse anti-human CD105 (Serotec, USA) were used for cell surface antigen detection, and all antibodies were conjugated with fluorescein isothiocyanate (FITC) or phycoerythrin (PE). Cells were stained for 30 min at 4°C. After antibody incubation, cells were washed with PBS and resuspended in 500 μl of PBS. Analysis was performed using FACS Calibur™ (BD Biosciences) and Cell Quest Pro™ (BD Biosciences) software.
Chondrogenic Differentiation
Chondrogenic differentiation was performed as previously described with some modifications (23). Briefly, 5 × 105 cells were added to a 15-ml polypropylene tube and centrifuged to a pellet. Chondrogenesis differentiation medium (Lonza) was added to the cell pellet at 37°C in a 5% CO2 incubator for 3 weeks. The differentiation medium was replaced three times per week. After 3 weeks, the round pellets were embedded in paraffin and cut into 3-μm sections. For histologic evaluation, the sections were stained with toluidine blue and alcian blue-PAS. A 3-μm cell pellet slice mounted on a slide was deparaffinized and hydrated with distilled water. For toluidine blue staining, the slide was immersed in toluidine blue working solution for 1 min. Excess unbound stain was removed by several washes using distilled water. The slide was quickly dehydrated with sequential washes of 95% and absolute alcohols. For alcian blue-PAS staining, the slide was stained with alcian blue (pH 2.5), periodic acid (0.5%), and Schiff's reagent. The slides were cleared in xylene and covered with Canada balsam and a cover slip.
Osteogenic Differentiation
Osteogenic differentiation was conducted with osteogenic medium, which was composed of 50 μM ascorbic acid 2-phosphate, 100 nM dexamethasone, 10 mM β-glycerophosphate (Sigma-Aldrich, USA), and 10% FBS in LG-DMEM. The basal culture medium was used as control medium. The differentiation medium was changed twice weekly for 3 weeks. After differentiation, the cells were stained with Alizarin Red S and Von Kossa to confirm calcium deposition. For Alizarin Red S staining, the cells were rinsed with PBS and fixed with 70% ice-cold ethanol for 1 h at 4°C. After three washes using distilled water they were stained with 40 mM Alizarin Red S (pH 4.2; Sigma-Aldrich, USA) for 10 min at room temperature. Nonspecific dye was removed by washing with distilled water five times. For Von Kossa staining, the cells were stained with silver nitrate (5%) for 30–60 min with exposure to ultraviolet light, followed by sodium thiosulfate (5%) for 2–3 min, and then counterstained with Nuclear Red Stain for 5 min. Alizarin Red S was solubilized in 100 mM of cetylpyridinium chloride (Sigma-Aldrich, USA) for 1 h. The release of the solubilized Alizarin Red S was measured at 570 nm using a spectrophotometer (18).
Adipogenic Differentiation
The adipogenic differentiation medium was composed of 500 μM 3-isobutyl-1-metyl-xanthine (IBMX), 1 μM dexamethasone, 60 μM indomethacin, and 5 μg/ ml insulin (Sigma-Aldrich, USA). Once cells reached 80–90% confluency, they were treated with the adipogenic differentiation medium for 3 weeks. To confirm adipogenic differentiation, Oil Red O staining was used. Cells were fixed with 10% formalin for at least 1 h and rinsed with 60% isopropanol prior to incubation in freshly diluted Oil Red O for 10 min. Bound stain was solubilized using 100% isopropanol, and the released stain was measured at 500 nm using a spectrophotometer.
Neurogenic Differentiation
The cells were seeded with the basal culture medium to a confluent population. Cells were preincubated for 24 h with 1 mM β-mercaptoethanol (BME; Sigma-Aldrich, USA) and 5% FBS. After preincubation, cells were incubated for 2 days in serum-free induction medium composed of 100 μM docosahexaenoic acid (DHA, Sigma-Aldrich, USA), B27 supplement (Gibco BRL, USA), and 1.5% dimethyl sulfoxide (DMSO, Sigma-Aldrich, USA) (9).
Immunostaining
For immunostaining, cells were fixed in 4% paraformaldehyde for 20 min, and were then permeablized for 10 min at room temperature in 0.5% Triton X-100 diluted in PBS. After washing three times, the cells were blocked with normal goat serum (NGS) overnight at 4°C. Cells were incubated with primary antibodies overnight at 4°C. After washing three times the cells were incubated with secondary antibodies Alexa 488 and Alexa 594 (1:1000, Molecular Probe, Inc., Eugene, OR, USA) for 1 h. Finally, for nuclear staining, Hoechst 33238 (1 mg/ml) was diluted 1:100 in PBS and loaded into samples for 15 min. Images were captured on a confocal microscope (Eclipse TE2000; Nikon, Japan). Primary antibodies used were rabbit anti-glial fibrillary acidic protein (GFAP, Millipore, USA) and mouse anti-neuron-specific βIII tubulin (TU-20, Abcam, UK).
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
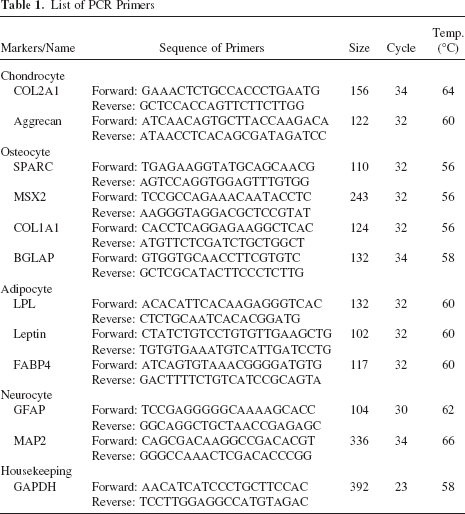
Total RNA was isolated from the cultured cells using TRIzol (Invitrogen, Carlsbad, CA). RNA concentrations were measured by absorbance at 260 nm with a spectrophotometer, and 1 μg of total RNA was used for reverse transcription using Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA). The cDNA was amplified using Taq Platinum (Invitrogen, Carlsbad, CA). The PCR primers are shown in the Table 1. PCR products were visualized with ethidium bromide on a 3% agarose gel.
List of PCR Primers
Statistical Analysis
The data were analyzed by Student's t-test using Excel software and expressed as the mean ± SE. Statistical significant data are indicated by asterisks (***p < 0.001, **p < 0.01).
Results
Isolation and Cell Culture of cWJ-MSCs
All samples of canine Wharton's jelly were acquired after cesarean section deliveries. The Wharton's jelly tissue is umbilical cord matrix, and the collected sample was moved to a sterile culture dish (Fig. 1). We isolated cWJ-MSCs from the Wharton's jelly using conventional methods for stem cell isolation and culture. Cultured cells were seeded in a T-25 cell culture flask with basal culture medium (LG-DMEM and 10% FBS), then collected and assessed. The morphology of the cells was fibroblast-like and spindle-shaped, which is similar to umbilical cord blood-derived MSCs (UCB-MSCs) and bone marrow-derived MSCs (BM-MSCs) (Fig. 1B). To determine stem cell ability and self-renewal capacity of cWJ-MSCs, we measured and calculated the proliferation via CPDL. The cells were cultured and maintained until passage 14. To measure the proliferation rate, 5 × 104 cells were seeded in a well of a six-well culture plate and subcultured 5–7 days later. We passaged the cells until we observed a decrease in the proliferation rate. A consistent increasing rate of cell proliferation through the cumulative population was observed (Fig. 1C). This result indicates that cWJ-MSCs have self-renewal capacity.

Primary culture of canine Wharton's jelly-derived mesenchymal stem cells (cWJ-MSCs) and identification of the cumulative population doubling level (CPDL). (A) Harvesting of canine Wharton's jelly tissue. (B) Phase contrast images of cWJ-MSCs. Cells were cultured with low glucose-Dulbecco's modified Eagle's medium (LG-DMEM) (10% fetal bovine serum; FBS). Cells exhibited fibroblast-like morphology and spindle shape similar to those displayed by human mesenchymal stem cells. Scale bar: 50 μm. (C) Cumulative growth curve of cWJ-MSCs. CPDL was evaluated with the formula described in Materials and Methods. The CPDL was measured from passage 3 to passage 14. Cells could grow consistently until passage 14.
Characterization of Immunophenotype by FACS Analysis
Generally, MSCs have distinct cell surface antigen markers. According to the International Society of Cellular Therapy, MSCs show positive expression for CD90 and CD105, and negative expression for CD11c, CD34, and CD45 other lineage negative surface antigen (3).
We conducted flow cytometry analysis of cWJ-MSCs at passage 3 and used 10 CD markers (CD3, CD11c, CD28, CD34, CD38, CD41a, CD45, CD62L, CD90, and CD105) to distinguish the MSC phenotype. Our results show an expression pattern consistent with the MSC phenotype (Fig. 2). cWJ-MSCs were stained positively for CD90 and CD105. CD90 is also called Thy-1 and is marker for various types of stem cells, such as hepatic stem cells, keratinocyte stem cells, endometrial stem cells, and mesenchymal stem cells (6,15,17). CD105 is also called endoglin and is a well-known MSC marker (3). Other markers for immune cells (CD3, CD11c, CD28, CD38, and CD62L), hematopoietic cells (CD34, CD45), and platelets (CD41a) were not detected. The FACS analysis results showed that cWJ-MSCs have a typical immunophenotype consistent with that of MSCs.
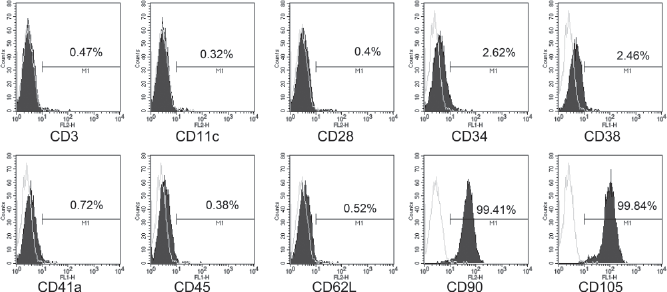
Fluorescence-activated cell sorting (FACS) analysis of cWJ-MSCs. Analysis was performed at passage 5. Values show the intensity of the indicated antigen.
Induction of Chondrogenic Differentiation
To confirm chondrogenesis, cWJ-MSCs were cultured with chondrogenic differentiation medium. The cells were seeded into 15-ml polypropylene tubes and centrifuged to make a pellet. The pellet was incubated in chondrogenic differentiation medium at 37°C in 5% CO2 for 2–3 weeks. The differentiation media was changed every 3 days. The pellet had an ovoid shape and an opaque body (Fig. 3A, B). The pellet formation is shown in the bottom of the tube (Fig. 3A). The pellet stained positively with toluidine blue (Fig. 3C) and alcian blue-PAS (Fig. 3D). We also validated the expression pattern of genes associated with chondrogenic markers such as collagen, type II, α1 (COL2A1), and aggrecan. The chondrogenic markers were increased under differentiation conditions compared with basal culture conditions (Fig. 3E).

Chondrogenic differentiation of cWJ-MSCs. After 3 weeks of chondrogenic induction, pellet formation was observed. (A) The pellet was formed at the bottom of 15-ml polypropylene tube. The black arrow indicates a pellet. (B) Image of an ovoid shaped chondrogenic pellet. (C) toluidine blue and (D) alcian blue-Periodic acid Schiff (PAS) staining of chondrogenic pellets. The pellets were embedded in paraffin and cut into 3-μm slices, which were mounted on a slide. Slides were stained with toluidine blue and alcian blue-PAS. The stained tissue showed a typical cartilaginous tissue phenotype. Scale bar: 100 μm. (E) RT-PCR for detection of mRNA expression level of chondrogenic-specific markers: collagen type II, α1 (COL2A1), and aggrecan. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as reference for evaluating the quality of mRNA.
Induction of Osteogenic Differentiation
To investigate osteogenesis, cWJ-MSCs were cultured in osteogenic induction medium. The differentiation medium was replaced every 3 days for 3 weeks. After differentiation, Alizarin Red S and Von Kossa staining were conducted to confirm osteogenesis. The basal culture medium was used as a control. Alizarin Red S and Von Kossa staining were positive under differentiation conditions (Fig. 4A, B, E, F). However, under the undifferentiation condition, the cells were negative after Alizarin Red S and Von Kossa staining (Fig. 4C, D, G, H). For quantification, the Alizarin Red S-stained cells were eluted with 100 mM of cetylpyridinium chloride. Osteogenic differentiated cells showed 10-fold greater values of Alizarin Red staining than control cells (Fig. 4I). When gene expression levels were examined, osteogenic differentiation markers, such as secreted protein, acidic, cysteine-rich (osteonectin; SPARC), msh homeobox homolog 2 (MSX2), COL1A1, and bone γ-carboxyglutamate (gla) protein (BGLAP) were expressed strongly under differentiation conditions (Fig. 4J).
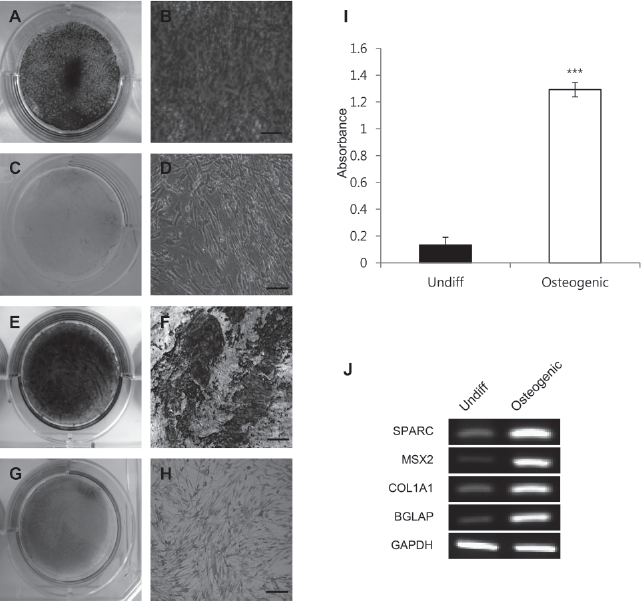
Osteogenic differentiation of cWJ -MSCs. (A–D) Alizarin Red S and (E–H) Von Kossa staining after 3 weeks of osteogenic induction. (A, B) Osteogenic differentiated cells were grown in osteogenic induction medium. Differentiated cells stained strongly with Alizarin Red S, which was distinct from control cells. Control cells were grown in normal LG-DMEM with 10% FBS. No staining with Alizarin Red S and Von Kossa was observed (C, D, G, H). Scale bar: 50 μm. For quantification, Alizarin Red S-stained cells were solubilized with 100 mM cetylpyridinium chloride, and the absorbance was measured spectrophotometrically at 570 nm for 0.5 s (I). Differentiated cells showed 13-fold greater staining than control cells. We performed all these analyses in triplicate and the mean ± SD is plotted (***p < 0.001). (J) RT-PCR for detection of mRNA expression levels of osteogenic-specific markers: secreted protein, acidic, cysteine-rich (osteonectin; SPARC), msh homeobox homolog 2 (MSX2), COL1A1, and bone γ-carboxyglutamate (gla) protein (BGLAP). GAPDH was used as reference for evaluating the quality of mRNA.
Induction of Adipogenic Differentiation
To demonstrate adipogenesis, cWJ-MSCs were cultured in adipogenic induction medium, which was changed every 3 days for 3 weeks. The basal culture medium was used as a control. Oil Red O staining was carried out to confirm adipogenesis. We detected fatty droplets under differentiation conditions, but not under control conditions (Fig. 5A–D). For quantification, stained cells were eluted with 100% isopropanol (Fig. 5E). The differentiated cells showed fivefold greater values than control cells. We also examined the gene expression levels of adipogenic associated markers such as lipoprotein lipase (LPL), leptin, and fatty acid binding protein 4 (FABP4). After differentiation, adipogenic-associated markers were increased compared to controls (Fig. 5F).

Adipogenic differentiation of cWJ-MSCs. (A–D) Oil Red O staining after 3 weeks of adipogenic induction. (A, B) Adipogenic differentiated cells were treated with adipogenic induction medium. Fat droplets in differentiated cells were stained by Oil Red O. The black arrow indicates a stained fat droplet. (C, D) Control cells were grown in the LG-DMEM with 10% FBS. There was no staining with Oil Red O. Scale bar: 50 μm. For quantification, stained cells were solubilized with 100% isopropanol, and absorbance was measured spectrophotometrically at 500 nm for 0.5 s (E). Differentiated cells showed fivefold greater staining than control cells. We performed all these analyses in triplicate and the mean ± SD is plotted (**p < 0.01). (F) RT-PCR for detection of mRNA expression levels of adipogenic-specific markers: lipoprotein lipase (LPL), leptin, and fatty acid binding protein 4 (FABP4). GAPDH was used as reference for evaluating the quality of mRNA.
Induction of Neural Differentiation
The cWJ-MSCs were cultured in the neural differentiation medium. The basal culture medium was used as a control. To confirm the neural differentiation, we performed immunostaining. The negative control, incubated with secondary antibodies, Alexa 488 and 594, demonstrated no background signal (Fig. 6A–C). We performed immunostaining for GFAP as a marker of astrocytes and βIII tubulin as a neural marker. Under basal culture conditions, cWJ-MSCs expressed GFAP, but not βIII tubulin (Fig. 6D, E). After neural differentiation, the cells expressed both GFAP and βIII tubulin (Fig. 6F, G). Therefore, we conducted that the relative populations of GFAP-positive and βIII tubulin-positive cells compared with control and neural differentiation condition. The quantification result showed that the relative populations of GFAP-positive cells similarly expressed both of them (control condition, 90.1%; neural differentiation condition, 93.2%) (Fig. 6H). βIII Tubulin-positive cells showed a positively relative population (9.2%) under neural differentiation condition (Fig. 6I). However, βIII tubulin-positive cells were not detected under control condition. When we examined gene expression, we found that GFAP was expressed under both control and neural differentiation conditions. Under differentiation conditions, microtubule-associated protein 2 (MAP2) was positive compared to control conditions (Fig. 6J). These data showed that cWJ-MSCs have the ability differentiate into neural cells.

Neural differentiation of cWJ-MSCs. cWJ-MSCs were immunostained with glial fibrillary acidic protein (GFAP) and βIII tubulin, astrocytes, and neural-specific markers. (A–C) Negative control was confirmed with Alexa 488 (B), Alexa 594 (C), and Hoechst only staining for nuclei detection (A). (D, E) Control cells were cultured with the LG-DMEM with 10% FBS. Control cells were positive for GFAP (D) but not for βIII tubulin (E). (F, G) After neural differentiation, differentiated cells were stained with GFAP and βIII tubulin. Scale bar: 50 μm. (H, I) The number of GFAP-positive and βIII tubulin-positive cells relative to the total nuclei cells was determined by counting and the mean ± SE is plotted (**p < 0.01). (J) RT-PCR for detection of mRNA expression levels of GFAP and microtubule-associated protein 2 (MAP2). GAPDH was used as reference for evaluating the quality of mRNA.
Discussion
In humans, there are many sources of stem cells, including bone marrow, umbilical cord blood, fatty tissue, and placenta (5,7,12,16,19), as well in specific organ tissue (10). Among these sources, there have been some reports of isolation and characterization of stem cells from umbilical cord matrix called Wharton's jelly in humans (14,26). Wharton's jelly is easy to obtain and is a plentiful source of stem cells. Moreover, Wharton's jelly is abandoned after delivery and classified as waste (26). However, little is known about stem cells from Wharton's jelly in canines. Here, we report the isolation and characterization of MSCs from canine Wharton's jelly for the first time. In our study, we isolated and cultured cells from four canine Wharton's jelly samples; the rate of success was 100%. All isolated cells (from four samples) showed similar cell morphology and ability of subculture. A single cell line was randomly selected. All experiments, CDPL, FACS analysis, and differentiation studies, were conducted by only the selected cell line (in triplicate). We cultured cWJ-MSCs with the basal culture medium (LG-DMEM with 10% FBS) for 14 passages. The cWJ-MSCs, like all stem cells, are capable of self-renewal. We demonstrated a robust cell proliferation of the cWJ-MSCs via calculation of cumulative population doubling level. The spindle-shaped morphology and adherent phenotype of the cWJ-MSCs is typical of mesenchymal stem cells (26). Immnuophenotype characterization indicated that cWJ-MSCs are positive for the mesenchymal stem cells markers CD90 (99.41%) and CD105 (99.84%). In hematopoietic surface makers, there showed negative or some expression pattern for CD45 (0.38%) and CD34 (2.62%). In the case of MSCs, usually, CD34 has a negative expression, but Viera et al. (27) showed 10% CD34-positive cells in canine adipose-derived mesenchymal stem cells, a larger positive population than ours. We suspect that canine stem cells might have some differences that were detected by the FACS analyses.
In order to confirm the mesenchymal ability of cWJ-MSCs, we conducted several differentiation studies. In the chondrogenic differentiation study, cWJ-MSCs showed the pellet formation and positively stained with toluidine blue and alcian blue-PAS under differentiation conditions. Also, the gene expression pattern of chondrogenic markers was increased under differentiation conditions compared to control conditions. After osteogenic differentiation, cWJ-MSCs showed abundantly positive staining with Alizarin Red S and Von Kossa. We quantified the expression levels of osteogenic marker genes, which were increasingly expressed after differentiation. We also found that cWJ-MSCs could be differentiated into adipogenic lineage cells. Under the adipogenic differentiation condition, some fatty droplets were stained with Oil Red O staining. As in other differentiation experiments, the gene expression levels of adipogenic markers increased after differentiation. In neural differentiation, we showed the expression of neural markers. cWJ-MSCs expressed GFAP, as shown in protein and gene levels, even under basal nondifferentiating conditions.
There are some reports using canine stem cells in regenerative medicine studies in dogs (1,22). Most of these reports indicate that the stem cells used originated from canine adipose tissue or umbilical cord blood (23,27). With regard to potential therapeutic use, an established diversity of stem cell source is needed.
In conclusion, our data showed that cWJ-MSCs can be easily isolated and can display the stem cell characteristic of self-renewal. The cWJ-MSCs can differentiate into chondroblasts, osteoblasts, adipocytes, and neural cells. Therefore, the cWJ-MSCs could have potential therapeutic properties for regenerative diseases and veterinary medicine. Also, we suggest that cWJ-MSCs will be useful for stem cell biological research and will be a good source of stem cells for preclinical fields in canines.
Footnotes
Acknowledgment
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST, 2010-0020265).