
Abstract
The use of human liver progenitor cells in the development of clinical cell therapy depends on their constant availability and unaltered properties during culture. The present study investigates the effects of long-term in vitro culture on the specific characteristics of these cells and on their genetic stability. Adult-derived human liver progenitor cells (ADHLPCs) were isolated from 12 donors and cultured until senescence and cell death. Cells were analyzed at different time points for their phenotype stability and differentiation potential. In addition, growth characteristics, chromosomal karyotype, telomere maintenance mechanisms, and activity of cell cycle-related genes were studied. Finally, their in vivo tumorigenicity was investigated in a xenograft assay. The long-term culture of ADHLPCs revealed a variable proliferation capacity. Cells maintained their original phenotype and acquired hepatocyte-like metabolic functions after differentiation. Eight of the 12 cell populations grew fast (doubling time of 6.3 days) during a limited time period (mean, 116.2 days), and mainly presented normal cytogenetic features. The four other cell cultures presented an early decline in growth potential (doubling time of 28.6 days) and premature senescence. Chromosomal alterations were detected in three of four cultures at passage 6. Cytogenetic anomalies were not correlated with tumorigenic potential in vitro or in vivo, and expression of cell cycle-related genes was appropriately upregulated, inducing senescence. Although chromosomal anomalies may occur in long-term cell cultures, neither transformation nor alteration of their characteristics was noted during in vitro expansion. All ADHLPCs reached senescence and growth arrest. Presenescent ADHLPCs might therefore be considered as a suitable source for liver-based cell therapy.
Introduction
Liver cell therapy has emerged as an attractive alternative to orthotopic liver transplantation for the treatment of liver inborn errors of metabolism. Clinical trials reported thus far have been performed using isolated mature hepatocytes (10,12). The limited cell availability, the low replicative potential of hepatocytes (10), and their loss of viability/function following cryopreservation (37) have restricted their use and given the impetus for further research on alternative cell sources. Our team and others have previously demonstrated that intra- and extrahepatic stem/progenitor cells have the potential to differentiate into hepatocyte-like cells both in vitro and in vivo following transplantation (6,17–20,25,33). Our team has demonstrated that adult-derived human liver progenitor cells (ADHLPCs) isolated from the liver parenchymal fraction show an advanced hepatic differentiation potential (16) when compared to skin fibroblasts (20) and bone marrow mesenchymal stem cells (19).
In order to supply sufficient cell mass to use in clinical development, cells need to be expanded in vitro. Recent studies have provided evidence that long-term ex vivo expansion of adult human mesenchymal stem cells originating from various tissues may alter their phenotype stability and/or induce spontaneous transformation (4,31,32,42). In these studies, cell transformation seemed to occur after a senescence phase (31) and to be associated with the disruption of cell cycle regulation (29). Reactivation of telomere maintenance mechanisms finally led to cell immortalization (13). However, this issue remains controversial (3,22,39), and some of these findings have since been retracted (8,40). Normal somatic human cells cultured in vitro have a limited life span (14) and enter senescence in response to different types of signals (35). A decline in proliferation and final growth arrest, known as replicative senescence, are triggered by gradual telomere shortening at each cell division. The accumulation of DNA damage related to in vitro culture conditions may also induce senescence. A disruption of cellular control checkpoints, via the inactivation of p53/p21 or pRb/p16, allows the bypassing of this senescence-related nonproliferative state. Cells continue to divide, although the short and dysfunctioning telomeres lead to general genomic instability, followed by general apoptosis, a phenomenon also referred to as the crisis phase. Human cells have rarely been documented to escape this fate and undergo transformation (42).
In the current study, we investigated the growth kinetics, phenotype, and genetic stability of ADHLPCs during long-term in vitro culture and consequently propose release criteria for cells aimed for replacement therapy.
Materials and Methods
Cell Isolation and Long-Term Culture
The present study was approved by the institution's ethical committee. The analyses were performed for preclinical testing purposes.
Human liver cells were obtained from liver segments of 12 healthy cadaveric donors (minimum age 9 years; maximum age 69 years; mean 25.62 ± 18.72 years) (Table 1) following size reduction of liver grafts. Cells were isolated and cultured to obtain adult-derived human liver progenitor cells (ADHLPCs) in line with a protocol described elsewhere (25). Cumulative population doublings (CPD) were calculated using the following equation: ∑[log10(NH)-log10(NI) / log10(2)], where NI is the inoculum number and NH is the harvested cell number. Bone marrow-derived mesenchymal stem cells were obtained from healthy cadaveric donors (19) after approval by the institutional ethical review board. Fibroblasts were obtained by skin biopsy from healthy donors (20) after written informed consent. HeLa cells were kindly provided by Prof. E. De Plaen (Ludwig Institute for Cancer Research, Brussels, Belgium). U2Os cells were kindly provided by Prof. F. Fuks (Laboratory of Cancer Epigenetics, ULB, Brussels, Belgium). Yeast strain YIG-397 was kindly provided by J-L. Gala (Center for human Genetics, UCL, Brussels, Belgium). Hepatoblastoma cell line HepG2 was purchased from ATCC. The experimental setup is outlined in Figure 1.

Experimental setup.
Characteristics of 12 Donors From Which Liver Cells Were Isolated

M, masculine; F, feminine; ND, data unavailable.
Adult-Derived Human Liver Progenitor Cell Characterization
Every three passages, ADHLPCs were analyzed for the expression of mesenchymal markers. To this end, cells were incubated for 25 min at 4°C with phycoerythrin (PE)-CD73, allophycocyanin (APC)-CD90 (both from BD Bioscience, Franklin Lakes, NJ, USA), fluorescein isothiocyanate (FITC)-CD105 (Ancell, Bayport, USA), or PE-CD133 (Miltenyi Biotec, Auburn, CA, USA) (following protocol described in Lysy et al. (20)) diluted in PBS containing 0.5% bovine serum albumin (BSA). Cell fluorescence was then measured using a fluorescence-activated cell sorting (FACS) Canto II flow cytometer and analyzed using the FACS Diva software. The corresponding isotypes (BD Bioscience) were used for evaluation of nonspecific binding. A minimum of 10,000 events were acquired for each sample. Immunofluorescence staining (6) was also performed to detect albumin (1/100, Sigma), vimentin (1/200, Sigma), α-smooth muscle actin (αSMA), as well as cytokeratin 18 and 19 expressions (1/200, Chemicon, Hants, UK). The nuclei were revealed with the 4′,6-diamidino-2-phenylindole nuclear dye (1/5,000; Sigma). The cells were examined with a fluorescent microscope (Axioscop, Zeiss).
Multilineage Differentiation Potential
Osteogenic, adipogenic, and hepatogenic differentiation was studied every three passages according to a protocol described elsewhere (6). Hepatic functional testing included a periodic acid Schiff staining, and an evaluation of urea production and cytochrome P450 3A4 (CYP3A4) metabolic activity (16). Hepatocytes and bone marrow-derived mesenchymal stem cells served as controls.
β-Galactosidase Staining
Senescence of cultured cells was evaluated using the senescence associated β-galactosidase (SA-β-gal) (15) activity at pH 6 staining kit (Sigma, St. Louis, MO, USA). After an overnight incubation with the reagents, cells were analyzed by optic microscopy (Leica) and photographed at 200x magnification. In total, 200 cells were counted for each experiment.
Limiting Dilution and Soft Agar Assays
Anchorage-independent growth and clonogenicity were assessed by culturing the cells in 0.3% soft agar (Agar Noble, Sigma) (5) and by plating a single cell into each well of a 96-well plate, followed by the monitoring of colony formation. Colonies of more than 10 cells were counted under optical microscope after 6 weeks. HepG2 cells served as inner positive control.
Telomerase Activity, Telomere Length Assay
Telomerase activity was assessed using the Trapeze Telomerase RT Detection kit (Chemicon, Brussels, Belgium). Normal and heat inactivated (85° for 20 min) HeLa cells were used as internal positive and negative controls, respectively. Telomere length was studied using the telo TAGGG Telomere Length assay kit (Roche, Vilvoorde, Belgium) according to the manufacturer's protocol. Genomic DNA was extracted from cells using the Genomic purification kit (Promega). U2Os (alternative lengthening of telomere-positive cells), HeLa (telomerase-positive cells), and young fibroblasts were used as internal controls.
Real Time Quantitative PCR and RT-PCR
Total RNA was extracted using the TriPure isolation reagent (Roche). RNA was quantified using a NanoDrop (Thermo Scientific, Waltham, MA, USA). cDNA was generated after DNAse treatment (Dnase kit Invitrogen) using the Thermoscript RT kit (Invitrogen). PCR amplification was performed using qPCR Mastermix SYBR Green I (Biorad, Nazareth, Belgium), cDNA made from 10 ng of RNA and the following set of primers at a final concentration of 300 nM: human telomerase reverse transcriptase (hTERT)-s CGGAAGAGTGTCTGGAGCA (LT5) and hTERT-as GGATGAAGCGGAGTCTGG (LT6). The experiment was performed in triplicate in an iCycler (Biorad) real-time PCR detection system. Human hepatocytes and HeLa cells served as internal negative and positive controls, respectively. Protocol for RT-PCR and primers used for hepatic nuclear factor (HNF)-4, albumin, cytokeratin 18, α-smooth muscle actin, and vimentin have been previously described (20).
FASAY (Functional Analysis of Separated Alleles in Yeast)
This assay was performed to evaluate the activity of the p53 gene using a previously described technique (11). Briefly, mRNA was extracted using the Dynabeads mRNA Direct kit (Dynal, Invitrogen. P53 mRNA was reverse transcribed, and exons 4–11 of the open reading frame of p53 were amplified by PCR. The yeast strain YlG-397 was then cotransformed with the p53 amplicon and a linearized yeast expression vector carrying the 5′ and 3′ ends of the p53 open reading frame. The yeast cells were grown onto low adenine plates. Because the mutant p53 is not able to activate its reporter, yeast cells grown under these conditions form red colonies, whereas cells containing the wild-type p53 form white colonies. At least 100 colonies were counted per sample. A total of less than 10% of red colonies were believed to be due to PCR-induced errors or the presence of an alternative spliced p53 mRNA and considered as a negative result (43). A FASAY assay resulting in 10–20% of red colonies was considered borderline. In these situations, a TP53 gene sequencing was performed to confirm or exclude p53 mutation. Cells derived from a human neck carcinoma with a well-characterized p53 mutation and from a Li-Fraumeni patient were used as positive controls.
Western Blot
Protein extracts were electrophoresed and transferred using standard methods. The blots were probed with mouse anti-cMyc (1/1,000, Cell Signalling, Danvers, US), mouse anti-p16 (1/1,000, Santa Cruz, Heidelberg, Germany), mouse anti-p21 (1/1,000, Calbiochem, Darmstadt, Germany), mouse anti-pRb (1/500, BD Bioscience), and mouse anti-β-actin (1/2,000, Sigma). Primary antibodies were revealed with a peroxydase-labeled goat antimouse antibody (1/10,000, Sigma). Blots were developed on a geldoc system (BioRad) using enhanced chemiluminescence (Amersham, Diegem, Belgium).
Molecular Karyotyping and Fluorescence In Situ Hybridization (FISH)
Cytogenetic analyses were performed (30) at passage (P) 4 in 6 of 12 cultures and systematically at P6. The cells were first blocked in metaphase by a 1-h incubation with 0.1 μg/ml of Colcemid, then subjected to hypotonic shock with 0.075 M KCl and fixed with a methanol/acetic acid (5/1 volume) fixative. The chromosomes were G-banded and subsequently examined by optical microscopy (Axioscop, Zeiss). A normal karyotype was defined as 200 metaphases counted and analyzed as normal by at least two investigators, in accordance with the International System for Human Cytogenetic Nomenclature (ISCN) 2009 (34). FISH was performed on interphase nuclei using the fluorescent-labeled DNA probe for TP53 gene (Vysis TP53, Abbott, Wavre, Belgium). The hybridization signal was analyzed on 100 intact nuclei by two investigators. A deletion was defined as the absence of one or both signals in >3% of nuclei.
In Vivo Tumorigenicity Assay
All animal experiments were performed in compliance with Belgian laws for animal protection and approved by the local ethical review board. To determine the tumorigenic potential of ADHLPCs in vivo, 107 cells were harvested at the end of P6, suspended in 300 μl of D-PBS, and injected subcutaneously into 5-week-old Balb-c NuNu mice (Charles River Laboratories, Wilmington, MA, USA). Cell viability was monitored before and after transplantation procedure using the Trypan blue dye exclusion method (Sigma) and propidium iodide/Annexin V (BD Bioscience) staining, followed by flow cytometric analysis, according to the manufacturer's protocol. Mice were monitored daily throughout the experiment and were euthanized when the tumor volume reached 0.5 cm3, or after 24 weeks, whichever came first. The skin and all the organs were harvested and fixed in 3.5% formaldehyde for further analysis.
Statistical Analysis
Statistical analysis was performed using Prism 4.1 (GraphPad software). Results are expressed as mean ± SEM. The probability of a significant difference between groups was determined using the unpaired t test. To address the question of multiple comparisons, the Bonferroni correction was used. Tests were considered to be statistically significant when ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05.
Results
ADHLPC Culture Characteristics
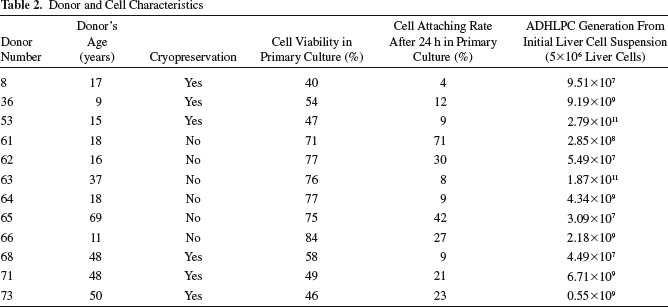
ADHLPC samples isolated from 12 different donors (Table 1) were used in the current study (characteristics are summarized in Table 2). Six samples (donors 61–66) were obtained from freshly isolated liver cell suspensions, while the other six (donors 8, 36, 53, 68, 71, and 73) were generated from cryopreserved/thawed liver cell batches. ADHLPCs were cultured in vitro up to senescence and death, corresponding to a period of more than 500 days. The long-term monitoring of cell growth revealed the presence of two different proliferative behaviors (Fig. 2A). Eight of 12 cell populations (named group A) had a fast growth, with a mean population doubling time (PDT) of 6.3 ± 2.7 days. These cell populations reached P6 after 101.3 ± 39.7 days and had a mean cumulative population doubling (CPD) of 14.8 ± 0.8. Among these, four populations (donors 36, 61, 64, and 66) reached replicative senescence, characterized by a decline in proliferation, at P7–8 corresponding to a CPD of 15.4 ± 2.1. The other four cell populations continued to proliferate until they reached a CPD of 15.5 ± 2.1 at P10 (donors 71 and 73) or a CPD of 22.1 ± 0.3 at P12 (donors 53 and 63). The second group (named group B), consisted of the remaining 4 of 12 populations (donors 8, 62, 65, and 68), grew slowly, with a PDT of 28.6 ± 17.4 days. In these populations, P6 was reached after 237.1 ± 27 days, corresponding to a CPD of 7.2 ± 0.2. A final CPD of 9.9 ± 0.4 was reached at P10–12 after more than 350 culture days. We were able to generate a mean of 4.58 × 1012 ADHLPCs in group A and 1.84 × 109 cells in group B from an initial cell suspension (5 × 106 liver cells) containing 5 × 104 stem cells (Table 2). No correlation was found between ADHLPC growth potential and the donor's age (pediatric versus adult). Indeed, the final CPD of cells originating from pediatric donors (less than 18 years) was 16.6 ± 1.3 as compared to 14.3 ± 2.3 for cells from adult donors (37–69 years) (p = 0.69). In addition, although thawed cells showed a lower initial adherence and viability when compared to freshly isolated cells (p < 0.001 and p = 0.12, respectively), this had no influence on ADHLPC growth potential. Indeed, there was no statistically significant difference in these two parameters between groups A and B (p = 0.96 for plating and p = 0.91 for viability, respectively) (Table 2), and the final CPD were similar for ADHLPCs issued from freshly isolated cell batches and ADHLPCs issued from thawed batches (14.6 ± 1.2 vs. 16.1 ± 3.3, p = 0.54). HepG2 cells served as a positive control and revealed an exponential proliferative capacity (Fig. 2B).

Growth kinetics, cell morphology, and senescence features during long-term ex vivo expansion of ADHLPCs. (A) Growth kinetics of ADHLPCs derived from 12 different donors defined as cumulative population doubling (CPD) over time in culture. ADHLPCs from cultures 8, 36, 53, 68, 71, and 73 were obtained after cryopreservation/thawing procedure of primary liver cell suspension, while the others resulted from fresh organ isolation. CPD potential varied between ADHLPC cultures. We distinguished two groups (A and B) based on the proliferative behavior. (B) Comparative growth curve of hepatoblastoma cell line HepG2. (C) Representative morphologic appearance of ADHLPCs at P3, P6, P9, and P12. Scale bar: 100 μm. ADHLPCs have a fibroblast-like morphology at early passages. Cells of group B acquired the flattened, irregular features of cell aging earlier than in group A. (D) SA-β-gal staining in groups A and B. The number of aging cells increased progressively during culture in both groups. Scale bar: 100 μm. (E) The percentage of senescent cells increased earlier on in ADHLPCs that grew slowly, presenting cytogenetic instability (group B) compared to cells with a good growth potential (group A).
Donor and Cell Characteristics
In parallel, we investigated the influence of culture age on cell morphology. Young ADHLPCs presented a fibroblast-like morphology, with a granular cytoplasm and an ovoid nucleus containing two or more nucleoli (Fig. 2C). With age, the cells progressively adapted a flattened, enlarged, and irregular appearance, their saturation density decreased, and the amount of cell debris in the culture medium increased. Changes in cell morphology appeared after P6–9 (mean CPD 15.2 for 186 ± 29.2 culture days) in the eight cell cultures of group A. This was associated with an average 63.7% positive SA-β-gal staining at P6, which increased to 93.2% at P9. The four cultures from group B presented morphological modifications and positive SA-β-gal staining (53.7%) early on, starting at P3–4 (mean CPD 5.1 for 107.8 ± 10.8 culture days) (Fig. 2D, E). Compared to fresh cultures, cells issued from thawed batches did not demonstrate earlier signs of senescence (data not shown).
Massive cell apoptosis, termed as the crisis phase, was never observed. None of the 12 cell cultures displayed the characteristics of transformed cells, such as loss of contact inhibition or small cell cluster formation.
ADHLPC Long-Term Culture Can Lead to Chromosomal Instability
We then studied the influence of long-term ex vivo culture conditions on ADHLPC genetic characteristics. The chromosomal stability of ADHLPCs was investigated during the exponential proliferation phase occurring and just prior to the senescence phase (P6–7). At P6–7, two of eight cell populations from group A and three of four cultures from group B showed significant numerical and/or structural genetic anomalies (summarized in Table 3), despite a normal karyotype at P4. We could not correlate the occurrence of cytogenetic alterations with CPD (p = 0.86). However, culture time seems to have an impact on karyotype findings (p = 0.016). Karyotype was performed after 224.8 ± 42.5 days in cells of group B. Cells of group A presenting cytogenetic instability (Fig. 3A) were analyzed after, respectively, 77 (donor 53) and 230 days (donor 63). The other cell cultures with a good growth potential had a normal karyotype (Fig. 3B). In those, cytogenetic analysis was performed after 101.3 ± 16.2 days. Interestingly, chromosomal aberrations were not identical between cultures.

Karyotype, TRF length, and telomere maintenance in ADHLPCs. (A) G-banded karyotype of donor 53 presenting a translocation [der(1)t(1;11)] in 14/50 mitosis. (B) Representative normal karyotype of an ADHLPC culture issued from group A. (C) Representative Southern blot analysis of telomere restriction fragment (TRF) of ADHLPCs at P6 issued from cells carrying a normal (lane 2) or abnormal karyotype (lane 3). Lane 1 represents the molecular weight marker. Telomere erosion is increased in the cells carrying genetic instability. Sarcoma cells U2Os displayed the typical long and heterogeneous TRF pattern of ALT-positive cells (lane 4). Young fibroblasts displayed long telomeres and did not demonstrate telomere erosion (lane 5). (D) Relative TRAP activity per microgram of protein lysate in ADHLPCs at P6 in cells with a normal (1) and abnormal karyotype (2). Telomerase-positive HeLa cells and enzyme thermo-inactivated (85°C during 10 min) HeLa cells (TI HeLa) served as inner positive and negative controls. TRAP activity was below the cutoff level for ADHLPCs. (E) Detection, by quantitative qPCR, of hTert m-RNA expression at P6 in ADHLPCs demonstrating the presence (E2) or absence (E1) of cytogenetic instability. HeLa cells served as internal positive control.
Results of Karyotypes Performed on ADHLPCs

[ ] Number of mitosis presenting described anomaly out of 50 mitosis counted.
Normal karyotype was defined as 200 normal mitosis. P, culture passage.
Cytogenetically Instable ADHLPCs Lack Telomere Maintenance Mechanisms but Present Telomere Erosion
In addition, we analyzed the telomere length of all cell cultures at P6 in order to investigate whether DNA instability might be associated with general telomere erosion. The telomere length was found to be similar in normal and cytogenetically instable cell cultures (4.9 kb). However, telomere erosion was more pronounced in the latter group (Fig. 3C), suggesting a potential reactivation of telomere maintenance mechanisms, a phenomenon involved in cell transformation. Therefore, all ADHLPC populations were tested for the activation of telomere maintenance mechanisms by functional telomere repeat amplification protocol (TRAP) assay and by qPCR for hTert transcription. Neither telomerase activity nor hTert expression could be detected in any of the cell populations at P3, 6, and 9 (Fig. 3D, E). In order to rule out the possibility of alternative lengthening of telomere (ALT) mechanisms, we also analyzed the telomere length heterogeneity by Southern blotting analysis of the telomere restriction fragment (TRF). We could not find any evidence of heterogeneity in ADHLPCs, in contrast with the ALT-positive U2Os cells (Fig. 3C).
Functional Maintenance of Cell Cycle Regulation Genes
The inactivation of tumor suppressor genes implied in cell cycle regulation has been shown to be involved in MSC transformation (29). We assessed by two complementary techniques the expression and functionality of the p53 tumor suppressor gene in all cell populations. The functionality of p53, as assessed by FASAY, was normal (less than 5% red colonies) for 10 cell populations at P6 (Fig. 4A). Cultures 36 (group A—with a normal karyotype, cryopreserved culture) and 62 (group B—presenting aneuploidy, fresh culture) showed a borderline FASAY, with 12% and 15% red colonies, respectively. DNA sequencing of the p53 gene was performed in those cells and ruled out the presence of a p53 mutation (data not shown). FISH on interphase nuclei of all ADHLPCs confirmed the maintenance (n = 100 nuclei) of both TP53 alleles on chromosome 17 (Fig. 4B). The analysis of p21, p16, and pRb expression by Western blotting revealed an increasing proteic expression of p21 and p16 with cell senescence, while pRb levels remained stable. Cells presenting chromosomal instability displayed an earlier upregulation of p21 and p16 levels, demonstrating the functionality of the cell cycle regulation machinery (Fig. 4C).

Tumor suppressors expression and in vivo tumorogenicity. (A) Representative results of FASAY experiment in cells derived from a neck carcinoma (A1) and in normal (A3) or cytogenetically unstable ADHLPCs presenting (A2). Histogram summarizing FASAY results for all cell cultures at P6 (A4). White colonies represent a wild-type p53; red colonies suggest a p53 mutation. The presence of a low percentage (<10%) of red colonies is due to the alternative splicing of the p53 gene or to copy errors during cDNA amplification. (B) Representative FISH analysis of ADHLPC interphase nuclei stained with TP53/CEN17 dual probe confirming the maintenance of both TP53 gene alleles in the 17p13.1 region. The green spot represents the TP53 locus, and the red spot corresponds to the centromere of chromosome 17. Scale bar: 15 μm. (C) Expression of p16, p21, and pRb proteins by Western blot analysis at P3 and P6 in ADHLPCs presenting (+) or not (-) chromosomal aberration.
Chromosomal Instability Is Not Associated with Tumorigenic Profile In Vitro
We further analyzed whether these karyotype abnormalities were associated with or could promote cell transformation. The clonogenic potential of all ADHLPCs was assessed in a limiting dilution assay. We never obtained a single-cell-derived clonal expansion in any of the cell populations, regardless of their karyotype. In contrast, HepG2 cells could easily give rise to clones (data not shown). Furthermore, normal cells require adhesion to a substrate before they can switch from a quiescent to a proliferative state. ADHLPCs, from any tested passage, were unable to form colonies on soft agar in contrast to HepG2 cells (Fig. 5A).

ADHLPCs do not display tumorigenic properties in vitro or in vivo. (A) ADHLPCs with (A1) or without (A2) cytogenetic instability could not proliferate on 0.3% soft agar in contrast with control HepG2 cells (A3). Top and bottom pictures represent the same experiment after 1 day and 6 weeks of culture, respectively. Scale bar: 200 μm. (B) Representative examples of Balb-c/Nu mice injected subcutaneously with 107 HepG2 cells or ADHLPCs. Tumor growth was observed macroscopically in mice injected 2 weeks earlier with HepG2 cells (B1). Representative mice injected with ADHLPCs did not demonstrate tumor development (B2).
ADHLPCs Did Not Induce Tumor Growth In Vivo
Finally, we investigated the effect of cytogenetic alterations on the in vivo behavior of ADHLPCs following subcutaneous transplantation in mice. Ten million ADHLPCs from each donor harvested at the end of P6 were injected into (n = 3) immunodeficient 5-week-old Balb-c NuNu mice. HepG2 cells were injected in the positive control group (n = 3). Cell viability was assessed after harvesting (92.1 ± 1.3%) and following injection (89.2 ± 2.8%) using the remaining cell fraction. When transplanted with HepG2 cells, all mice demonstrated tumor growth and general signs of illness (anorexia, cachexia, lordosis) 2 weeks after transplantation (Fig. 5B-1). In contrast, mice transplanted with normal and abnormally karyotyped cells did not show any sign of tumor formation after a 24-week monitoring (Fig. 5B-2).
Phenotypic Characteristics of ADHLPCs in Long-Term Culture
The expression of ADHLPC surface markers was analyzed by flow cytometry every three passages (Fig. 6A). The cells expressed the mesenchymal lineage markers CD73 (30 ± 12.5) and CD90 (90 ± 12) but were negative for CD105 (5.8 ± 3.1). The expression of cytoplasmic markers was assessed using immunofluorescence and PCR (Fig. 6B, C). Mesenchymal affiliation was confirmed, as the cells consistently expressed vimentin and αSMA. Positive staining for albumin and HNF-4 attested the hepatic origin of ADHLPCs. The cells did not express hepato-epithelial markers such as cytokeratin 18 or the oval cell marker cytokeratin 19 (data not shown). A stable immunophenotype was observed in all samples until cell senescence.

Phenotypic characterization of ADHLPCs. (A) FACS assay performed on n = 12 ADHLPCs showing the expression levels of CD73, CD 90, CD105, and their stability between P3 and 9. ADHLPCs were incubated with PE-CD73, FITC-CD105, or APC-CD90 monoclonal antibodies or matched isotype controls. (B) Representative immunofluorescence staining of (n = 12) ADHLPCs for hepatocyte and mesenchymal markers. Cells expressed albumin, α-smooth muscle actine (αSMA), vimentin, and cytokeratin 18. Hepatocytes and bone marrow-derived MSCs served as positive controls. Scale bar: 100 μm. (C) RT-PCR analysis revealed the stable expression of HNF-4, albumin, αSMA, and vimentin in ADHLPCs at P3, P6, and P9. Cytokeratin 18 was not detected in ADHLPCs. Human hepatocytes (hH) were used as positive controls.
Because CD133 was recently identified as a tumorigenic stem cell marker (21), its expression was also assessed. We could not detect any expression of CD133 (0.2–0.5%) in either normal or abnormally karyotyped ADHLPCs at P3, 6, 9, or 12 (data not shown).
Differentiation Potential of ADHLPCs
One of the characteristics of ADHLPCs is their potential to differentiate into hepatocyte-like cells. We assessed how culturing ADHLPCs until senescence could affect their ability to differentiate along the hepatic lineage. ADHLPCs from P3, 6, and 9 were cultured in hepatocyte differentiation medium. Cells of both groups—with respect to growth potential—could morphologically differentiate into hepatocyte-like cells up to P6. Indeed, cells acquired a polygonal shape and contained enhanced cytoplasmic granulations and a central nucleus. At P9, the cells maintained their initial spindle shape morphology despite the differentiation stimuli, suggesting that the differentiation capacity of these cells is limited by aging. These morphological findings were correlated to mature hepatocyte-related functions. The periodic acid Schiff staining showed that ADHLPCs that could morphologically differentiate also displayed the ability to store glycogen. CYP3A4 activity, and urea production were also upregulated in those differentiated ADHLPCs but decreased at P9 (Fig. 7). Cryopreservation procedures did not affect hepatocyte-like differentiation potential of ADHLPCs (data not shown).
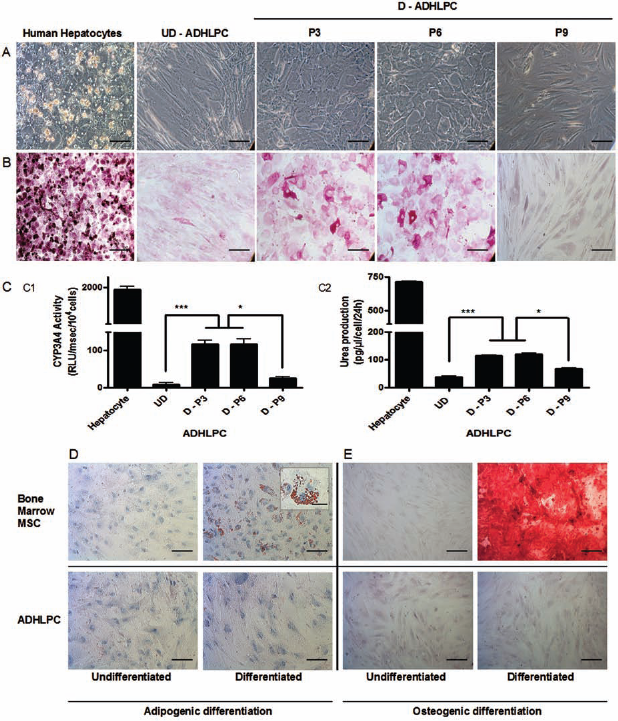
Osteogenic, adipogenic, and hepatocyte-like differentiation of ADHLPCs. (A) Hepatocyte-like differentiation capacity of ADHLPCs at different culture passages observed by phase-contrast microscopy. (B) The glycogen storage capacity was tested by periodic acid Schiff staining. Scale bar: 100 μm. Adult hepatocytes served as positive control. Undifferentiated (UD)—ADHLPCs cultured in medium without hepatogenic supplements. Differentiated (D)—ADHLPCs cultured in hepatogenic differentiation medium at P3, 6, and 9 respectively. (C) CYP3A4 activity (C1) and urea production (C2) in undifferentiated (UD) and hepatocyte-like differentiated (D) ADHLPCs at P3, 6, and 9. ADHLPCs demonstrating morphological hepatocyte-like differentiation showed mature hepatocyte functions confirmed by a positive acid Schiff staining, the capacity to produce urea and CYP3A4 activity. Cells that were cultured in unsupplemented medium and senescent cells unable to differentiate morphologically did not display these characteristics. Data are represented as means ± SEM. ∗∗∗p ≤ 0.001; ∗0.01 ≤ p < 0.05. (D–E) ADHLPCs, here shown at P6 (representative for P3 and P9), did not exhibit adipogenic (D) or osteogenic (E) differentiation potential, in contrast with bone marrow MSCs. Bone nodules were revealed by Alizarin Red staining, while lipid droplets positively stained for Oil Red O. Scale bar: 100 μm, inset scale: 50 μm.
As expected, none of the ADHLPCs had the ability to differentiate into the osteoadipogenic lineages as confirmed by the absence of bone matrix deposition colored by Alizarin Red and the lack of lipid droplets, as emphasized by Red Oil O staining (data not shown).
Discussion
This study addresses an important and frequently raised issue on the safety of human regenerative medicine using in vitro expanded cells. We therefore analyzed the phenotype and genotype stability of ADHLPCs derived from 12 different liver donors during long-term in vitro culture. The cells never displayed uncontrolled proliferation or signs of transformation. Karyotypic abnormalities always led to the induction of senescence.
In vitro cell expansion is commonly used in cell therapy in order to reach the appropriate cell mass necessary for clinical use. During the in vitro culture, cells taken out of their natural niche undergo multiple divisions and are exposed to different culture-related stresses that might induce premature cell senescence and chromosomal instability (35).
The human liver progenitor cells used in this study could be expanded in vitro up to 15.2 ± 4.2 PDs over more than 300 days. Compared to fibroblasts (60–80 doublings) (14) and bone marrow-derived MSCs (20–40 doublings) (2), ADHLPCs had a lower proliferation potential which might be related to their progenitor status. In line with previous works, we showed that ADHLPC proliferation kinetics differed between donors (3,22). The fact that the initial cell suspension had been cryopreserved did not influence cell growth potential. Although an older donor's age is reported to influence cell growth potential (22,36), we did not observe any age-related differences among cell populations. However, this discrepancy could be explained by the fact that our patients were younger than in the study of Stenderup et al. (36) (age between 66 and 81 years). The number of senescent cells increased gradually with the length of in vitro cell culture; interestingly, this phenomenon appeared earlier in the cell populations, which grew slowly. Unphysiologic high oxygen levels (21%) during culture are also known to alter cell growth potential, and senescence might be delayed by using lower oxygen concentrations (38).
We demonstrated, in line with other authors, progressive morphological changes during cell culture. Although, in contrast to others studies, the original phenotypic properties were maintained until cell death in all ADHLPC samples (1,4,41). Cell cryopreservation did not alter their characteristics. The in vitro hepatocyte differentiation capacity also remained stable for both ADHLPC groups up to P6 (p = 0.55) after which it became impaired for all donors. These results are in line with similar observations made for senescent bone and adipose tissue-derived mesenchymal stem cells (1,4,41).
Structural and numerical chromosomal abnormalities were observed in our study in 5 of 12 ADHLPC samples. The anomalies found were different for the five samples, excluding the hypothesis of a localized chromosomal fragility linked to the culture conditions. The cell cultures presenting chromosomal instability at P6 had a normal karyotype at P4, suggesting that the alterations were acquired as a result of long-term culture. Recent studies question the risk for stem cell transformation during long-term ex vivo culture. Although human stem cells seem more resistant to transformation than murine-derived cells (24), Wang and others (31,32,42) have demonstrated that human bone marrow mesenchymal stem cells sometimes undergo transformation. Some of these findings were however later retracted due to cross-contamination (8,40). On the other hand, MSCs studied by Bernardo et al. (3) remained genetically stable over the same culture period. General culture conditions and duration are known to influence genome stability (35). Although all cells were cultured in the same conditions, by the same operator and using the same batch of serum, P6 was reached after different culture times and cumulative population doubling. Karyotyping was performed after 224.8 ± 42.5 days in abnormally behaving cultures, and after 101.3 ± 16.2 days of culture in normal ones (p = 0.016) with the exception of the culture originating from donor 53, presenting cytogenetic instability after 77 days in culture. Recurrent cytogenetic instability detected in culture 53 after a relatively short culture period evoked a donor-related susceptibility, a fact already underlined by Tarte et al. (39). A donor's age and lifestyle (smoking, diet, toxic exposure) are factors that have been described as potentially increasing cytogenetic damage (9,27). Unfortunately, we were not able to gather enough information on the donors to investigate these variables. Telomere erosion was more pronounced in cytogenetically unstable cells than in other cultures. During long-term culture, cells are exposed to oxidative stress, which is known to induce DNA damage and accelerate telomere erosion (7,26). In normal culture conditions, cells are grown in 21% oxygen, which is a level above the physiological values (3–5%) observed in the different organs of the organism. Sensitivity to oxidative stress induced DNA damage differs between individuals (28). This process takes time to settle double-stranded breaks and may explain the DNA instability found in cells that grew slowly for a prolonged period of time.
Whether or not cultured cells carried chromosomal abnormalities, in vitro and in vivo assays did not reveal any tumorigenic potential. All cell populations showed limited replicative potential and failed to show any sign of a crisis phase. ADHLPCs in long-term cultures did not express telomerase maintenance mechanisms, a phenomenon associated with MSC transformation in Wang et al.'s report (42). We could not detect mechanisms of alternative lengthening of telomeres nor hTert RNA transcript expression. In contrast with cancer cells, ADHLPCs did not grow on soft agar or expressed contact inhibition mechanisms. Several attempts to obtain single cell-derived clones failed and ADHLPCs could not give rise to tumor growth in vivo. In agreement with previous reports, chromosomal anomalies sometimes occurred but did not confer a survival advantage to the cell population (23,39). On the contrary, they seemed to induce cell senescence. Functionality and expression of p53, pRb, p16, and p21, major keys in cell cycle regulation, increased with aging in all ADHLPC cultures but were upregulated earlier in those presenting cytogenetic instability. This suggests that cells carrying karyotypic instability demonstrate the capacity to regulate their cell cycle and induce premature senescence as a potent mechanism against transformation.
The expression of the endothelial marker CD133, which has been reported on liver cancer stem cells (21), remained negative in all ADHLPC samples during the whole culture process.
In conclusion, our data show that ADHLPCs cultured in vitro for an extended period presented a variable proliferation capacity. Cells cultured for a prolonged period of time sometimes displayed cytogenetic instability that could not be correlated with a transformed behavior in vitro or in vivo. Cancer is a complex multistep process, and ADHLPCs exhibit multiple gatekeepers to prevent cell transformation. In this context, the monitoring of genetic characteristics plays an important role in the early detection of premalignant states. Furthermore, we suggest to limit culture period to guarantee the release of safe cell batches for patient therapy.
Footnotes
Acknowledgments
The authors are grateful to Christine Sempoux and Amandine Van Beneden for their excellent technical assistance with fluorescent microscopy and TRF assay, respectively, to Floriane Andre and Nawal Jazouli for excellent technical coaching. Thibault Helleputte for performing statistical analysis. This work was supported by Grant Waleo (Hepatera) from the Region Wallone, grant FNRS No. 7.4592.07 and by the Fondation Saint Luc. The authors declare no conflicts of interest.