
Abstract
Presently, orthotopic liver transplant is the major therapeutic option for patients affected by primary liver diseases. This procedure is characterized by major invasive surgery, scarcity of donor organs, high costs, and lifelong immunosuppressive treatment. Transplant of hepatic precursor cells represents an attractive alternative. These cells could be used either for allogeneic transplantation or for autologous transplant after ex vivo genetic modification. We used stromal cells isolated from adipose tissue (AT-SCs) as platforms for autologous cell-mediated gene therapy. AT-SCs were transduced with lentiviral vectors expressing firefly luciferase, allowing for transplanted cell tracking by bioluminescent imaging (BLI). As a complementary approach, we followed circulating human α1-antitrypsin (hAAT) levels after infusion of AT-SCs overexpressing hAAT. Cells were transplanted into syngeneic mice after CCl4-induced hepatic injury. Luciferase bioluminescence signals and serum hAAT levels were measured at different time points after transplantation and demonstrate persistence of transplanted cells for up to 2 months after administration. These data, along with immunohistochemical analysis, suggest engraftment and repopulation of injured livers by transplanted AT-SCs. Moreover, by transcriptional targeting using cellular tissue-specific regulatory sequences, we confirmed that AT-SCs differentiate towards a hepatogenic-like phenotype in vitro and in vivo. Additionally, in transplanted cells reisolated from recipient animals' livers, we detected activation of the α-fetoprotein (AFP) promoter. This promoter is normally transcriptionally silenced in adult tissues but can be reactivated during liver regeneration, suggesting commitment towards hepatogenic-like differentiation of engrafted cells in vivo. Our data support AT-SC-mediated gene therapy as an innovative therapeutic option for disorders of liver metabolism.
Introduction
Inherited diseases of liver metabolism, which are mostly caused by deficiency of hepatic proteins, together with chronic liver degenerating diseases, are a serious healthcare problem. At present, orthotopic liver transplantation (OLT) represents the ultimate treatment for these diseases. However, clinical application of this surgical procedure has some limitations including organ scarcity, major surgical intervention, and lifelong immunosuppression, which often demonstrates complications, high costs, and eventual organ rejection (8). Hepatocyte transplant (HcTx) represents an alternative procedure to liver transplantation (13). One of the main problems encountered in the practice of HcTx is the availability of livers for cell isolation. Hepatocytes are generally obtained from organs not considered suitable for liver transplantation, affecting the quality of isolated cells and limiting the possibility of their cryopreservation. Another major hurdle of HcTx is the short-term clinical efficacy, due to the limited repopulation capability of hepatocytes. This limitation may arise from the terminally differentiated status of the transplanted cells (13). Despite these problems, HcTx has proved to be effective in Crigler–Najjar disease (10) and α1-antitrypsin and ornithine transcarbamylase deficiency (43) even when transplanted cells make up only 1–5% of the total hepatocyte mass. These studies provide proof of principle for cell transplantation as a valuable therapeutic option for several inherited liver metabolic diseases.
The cell therapy approach for liver disease was aided by the identification of precursor cells able to differentiate into hepatocytes (18,42). In rodents, both hepatocytes and biliary epithelia arise from rare hepatic progenitors (oval cells) present in the liver near the canals of Hering, but the precise role of such cells in liver regeneration is unclear (45). Furthermore, isolation and amplification of hepatic progenitors cells from human liver samples are challenging since their exact localization and expression of specific markers are not completely understood (33). However, hepatocyte-like cells have been obtained from cells isolated from a growing number of extra hepatic tissues [for review, see (18)], including adipose tissue (2,30,38). Clinical use of hepatic precursor cell transplant has been proposed as a bridge to OLT (28), performed on a Crigler–Najjar patient (15) and for the treatment of liver cirrhosis (32); however, translational research on this topic is incomplete (33).
Adipose tissue (AT) represents an attractive cell source for the development of cell therapy and cell-mediated gene therapy platforms (12). AT is abundant, replenishable, and accessible with minimal invasive procedures. When subjected to specific stimuli, adipose tissue-derived stromal cells (AT-SCs) are able to differentiate into several cells types of both mesodermal and nonmesodermal origin, including hepatocytes [for review, see (34)].
In the current study, we show that genetically modified AT-SCs administered into an animal model of hepatic injury may engraft and repopulate injured liver with concomitant expression of a therapeutic transgene. By in vivo bioluminescent imaging (BLI), we were able to track administered cells and determine their biodistribution and persistence for up to 2 months after infusion. Transplanted cells predominantly engraft in regions undergoing liver regeneration and acquire some markers specific of a hepatic phenotype. In addition, transplanted cells can be reisolated from the liver of recipient animals. In these cells, we detected activation of the α-fetoprotein (AFP) promoter, suggesting commitment towards hepatogenic differentiation in vivo (17).
Taken together, our data indicate that AT-SCs may represent a valuable supply of hepatic precursors as well as a suitable target for cell-mediated gene therapy for liver disease.
Materials and Methods
Cell Isolation and Culture
AT-SCs were obtained from 6-week-old wild-type Swiss CD1 mice as previously described (6). Approximately 0.8–1.2 × 106 AT-SCs were isolated from each 6-week-old mouse (5). Fluorescence-activated cell sorting (FACS) analysis at second passage confirms the expression of typical adipose tissue-derived mesenchymal stromal cell (AT-MSC) markers such as cluster of differentiation 90 (CD90) and CD54 and the absence of hematopoietic and endothelial markers such as CD45, CD34, and CD31. Human hepatocarcinoma HEP G2 (ATCC® No. HB-8065) and human adenocarcinoma HeLa (ATCC® No. CCL-2) cells were cultured following ATCC recommendations.
In Vitro Hepatogenic Differentiation
AT-SCs were used for differentiation assays according to a protocol originally described to induce in vitro a hepatic phenotype in of human bone marrow mesenchymal cells (20). A method to induce hepatic differentiation in mesenchymal cells isolated from adipose tissue was subsequently described by Talens-Visconti et al. (44). Briefly, freshly isolated adipose tissue cells were plated at 2.5 × 104 cells/cm2 on tissue culture dishes in α-modified minimum essential medium (α-MEM) supplemented with 20% fetal bovine serum (FBS), 2 mM l-glutamine, and 1% penicillin–streptomycin and incubated overnight at 37°C and 5% CO2 (preselection step). Nonadherent cells were discarded, medium was replaced, and cells were allowed to grow until they reached 80% confluence (expansion step). Cells were then subcultured at 1:3 ratio, and starting the next day, they were serum-deprived for 2 days in α-MEM. Cells were then cultured in the same medium supplemented with 20 ng/ml epidermal growth factor (EGF) and 10 ng/ml basic fibroblast growth factor (bFGF) for 7 days (conditioning step). Induction of hepatic phenotype was achieved by sequential addition of 20 ng/ml hepatic growth factor (HGF), 10 ng/ml bFGF, and 4.9 mmol/L nicotinamide, for 7 days (differentiation step); then α-MEM was supplemented with 20 ng/ml oncostatin M (OMS), 1 μmol/L dexamethasone, and 10 μl/ml insulin transferrin selenium (ITS) + premix for 5 more days to achieve cell maturation (terminal differentiation step).
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from cells at different time points during the hepatic differentiation procedure using an RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. RNA was reverse-transcribed with Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega, Madison, WI) using random hexamers. Thermal cycling conditions and primers used for RT-PCR of liver-associated genes have been described elsewhere (36,39).
Lentiviral Vector Production and Titration
Lentiviral vectors (LVs) used in this study were third-generation self-inactivating (SIN) vectors derived from pCCLsin.cPPT.hPGK.E-GFP.Wpre (phosphoglycerate kinase promoter-enhanced jellyfish green fluorescent protein) (9) obtained from E. Vigna (IRCC, Candiolo, Italy). We generated LVs containing the ubiquitously active PGK promoter followed by either firefly luciferase (fluc) cDNA or human α1-antitrypsin (hAAT) cDNA as replacements to E-GFP cDNA. For transcriptional targeting studies, we generated a LV expressing a marker under the control of the liver-specific α-fetoprotein promoter (AFP). This vector was derived from the PGK.E-GFP plasmid by substituting the 0.5-kb human PGK promoter with the 2.1-kb human α-fetoprotein enhancer and promoter derived from pDrive AFP-hAFP (Invivogen, San Diego, CA).
Recombinant vesicular stomatitis virus-pseudotyped LVs were obtained as previously described (9). Titers of GFP-expressing LV stocks were determined by serial dilution on HeLa cells and flow cytometry analysis and were above 1 × 108 transducing units/ml (TU/ml).
Lentiviral-Mediated Gene Transfer Into AT-SCs
Second passage cells were used for lentiviral-mediated gene transfer. AT-SCs were plated at 60% confluence in α-MEM supplemented with 20% FBS. On the next day, cells were transduced with 10 TU/cell of LV stocks in the presence of 6 μg/ml Polybrene®. After incubation at 37°C overnight, cells were trypsinized and subcultured at 1:3 ratio and cultured for 2 more days. Cells were then harvested using trypsin/EDTA, washed in PBS, and used for FACS analysis and transplantation. Transduction efficiency was attested to be above 90% in all experiments. Infectivity of LVs decays logarithmically with time; the estimated half time of the LVs in a cell-free environment at 37°C is less than 10 h (51). However, to exclude any contaminating viral particles in the transplantation solution, 0.45-μm filtered LV-E-GFP cell supernatant was used to transduce HeLa cells. Neither FACS analysis nor fluorescent microscopy revealed any E-GFP-positive cells, excluding the presence of active viral particles in the samples.
AT-SCs Transplant
Eight-week-old wild-type Swiss CD1 mice were used in experimental groups of at least six animals. All experimental procedures were performed according to the guidelines of the Italian National Institutes of Health and were approved by the institutional animal care and use committee.
Acute hepatic injury was induced by intraperitoneal injection of carbon tetrachloride (CCl4; 1 ml/kg) (Sigma-Aldrich, St. Louis, MO) dissolved in peanut oil (27). On the following day, genetically modified AT-SCs (2.5 × 105 in 100 μl PBS per mouse) were administered into the spleen using a 27-gauge needle, as previously described (29). Then the abdominal wall was sutured, and the skin was closed with wound clips.
Ex Vivo and In Vivo Optical Bioluminescence Imaging
Ex vivo and in vivo imaging analysis was performed using the IVIS® Lumina equipped with Living Image 3.1 software (Caliper Life Sciences, Hopkinton, MA). For ex vivo imaging, cells were plated into clear bottomed tissue culture dishes and incubated in a solution of d-luciferin (Caliper Life Sciences) dissolved in tissue culture medium (150 μg/ml) before analysis. For in vivo analysis, mice were anesthetized with Avertin® and d-luciferin dissolved in PBS (150 mg/kg body weight) was administered IP 10 min before analysis. Images were recorded between 1 and 5 min, depending on the intensity of the bioluminescence emission. The same mice were analyzed at different time points after transplant. Necropsy was performed, and single organs were analyzed ex vivo. Luciferase activity in cell and tissue homogenates was detected with a specific assay (Promega, Madison, WI).
Quantification of Human AAT Levels
Blood samples were obtained via retro-orbital bleeding under sedation. Serum was obtained by centrifugation at 1,500 × g for 10 min. Serum levels of hAAT were determined using the human AAT ELISA Kit from Immundiagnostik AG (Bensheim, Germany).
Immunohistochemistry
Immunohistochemistry was performed on 5-μm consecutive sections of formalin-fixed, paraffin-embedded tissue with antibodies against firefly luciferase (Sigma Aldrich) and murine GATA4 (Novus Biologicals, Littleton, CO).
Statistical Analysis
Data analysis and comparisons between control and treated groups were done with INSTAT (GraphPad, San Diego). The significance of differences was assessed with a two-tailed Student's t test for unpaired data; statistical significance level was set at p < 0.05. Results are expressed as means ± SEM.
Results
Transdifferentiation Potential and Gene Transfer Into AT-SCs
Several reports have proved that human AT-derived MSCs undergo in vitro differentiation towards the hepatogenic phenotype in the presence of selective inductive media (2,3,20). Our data indicate that murine AT-SCs derived from inguinal fat pads also have hepatogenic transdifferentiation proprieties. Cytomorphological changes observed during the differentiation protocol were associated with temporal expression of liver-specific transcripts, including GATA4, AFP, albumin, cytochrome P450 (CYP2B), and cytokeratin-18 and −19 (CK-18 and CK-19) as assessed by RT-PCR analysis (Fig. 1A).
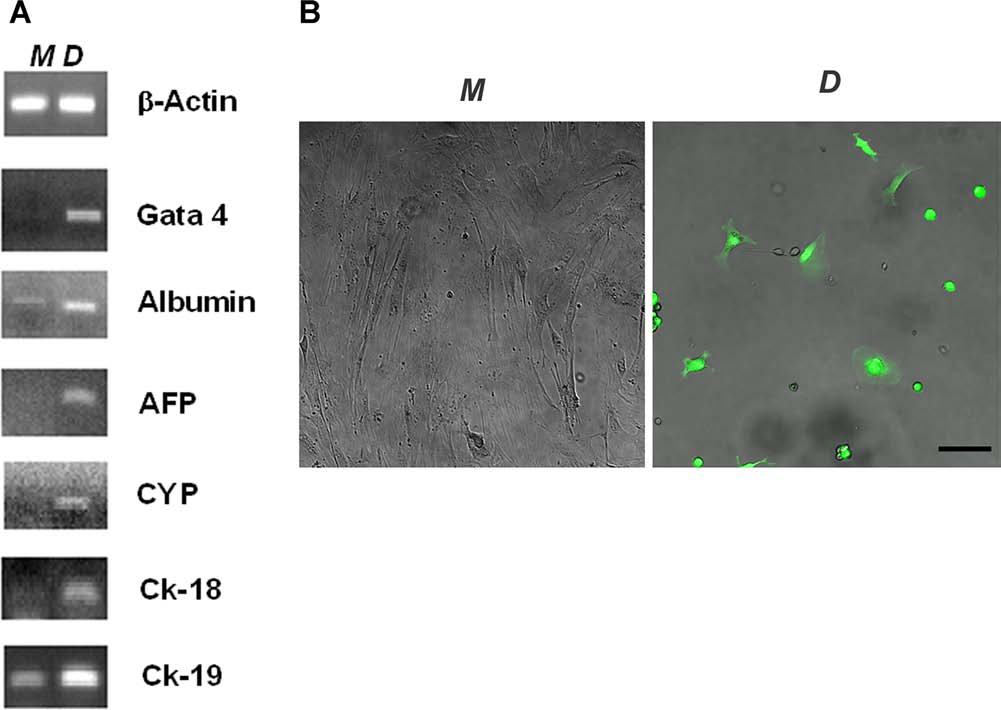
In vitro transdifferentiation potential of AT-SCs. Gene transfer into adipose tissue derived stromal cells (AT-SCs) was performed using a lentiviral vector (LV) bearing an expression cassette driving enhanced green fluorescent protein (E-GFP) under the control of the liver-specific α-fetoprotein (hAFP) promoter. Second passage AT-SCs were induced to differentiate towards a hepatogenic phenotype in a multistep, 3-week-long protocol. To determine whether differentiated cells show characteristic expression of a hepatic phenotype, expression of liver markers was determined by RT-PCR (A) in cells expanded in maintenance (M) or differentiating medium (D). AT-SCs cultured in maintenance medium showed a higher proliferation rate compared to AT-SCs cultured in differentiating medium. Human AFP promoter-specific E-GFP expression was detectable by fluorescent microscopy analysis only in cells cultured in hepatogenic differentiation (D) medium (B). Scale bar: 100 μm. CYP, cytochrome p450; CK-18, cytokeratin-18.
Potential activation of a hepatocyte-like expression profile in AT-SCs was also confirmed by liver-specific transcriptional targeting with a fluorescent marker. During embryogenesis (6–7 days of murine gestation), the AFP gene is normally expressed in visceral endoderm of the yolk sac (19). At a later developmental stage, maximal expression is observed in fetal liver and, at significantly lower levels, in embryonic gut. AFP is transcriptionally silenced in adult tissues but can be restored during liver regeneration and in primary hepatic tumors (17,26). Activation of the AFP promoter in the liver is highly specific, being regulated by a small set of transcription factors including hepatocyte nuclear factor-1, −3, and −4 (HNF-1, HNF-3, HNF-4) and CCAAT/enhancer binding protein (C/EBP), which share a common liver-restricted tissue distribution (24). HNF-1 and HNF-4 are known to play a crucial role in the determination and maintenance of hepatocyte-specific differentiation (7).
We tested the susceptibility of AT-SCs to lentiviral-mediated gene transfer. A lentiviral vector (LV) expressing E-GFP under the control of the human AFP enhancer and promoter was generated. Specific hAFP promoter-mediated expression of E-GFP was assessed in vitro in HEP G2 cells, reported to express high levels of AFP, while it was undetectable in AFP-negative HeLa cells (data not shown). The LV expressing E-GFP under the control of the AFP enhancer/promoter was then used for gene transfer into second passage AT-SCs. LV-treated cells were then cultured in either transdifferentiation medium as described above or maintenance medium. Differentiation into the hepatic-like phenotype in AT-SCs was associated with E-GFP expression (Fig. 1B, right). After 10 days in differentiation medium (first differentiation step indicated in Materials and Methods section), approximately 40–50% of AT-SCs turn the AFP promoter on (express E-GFP), while after 3 weeks (terminal differentiation step) the percentage drops to 20–30%. This is possibly due to the fact that, as described in the literature, AFP expression is turned off in some cells during the terminal differentiation stage. Cells maintained in α-MEM 20% FBS did not express E-GFP (Fig. 1B, left), indicating an activation of the fetal liver-specific AFP enhancer/promoter only in hepatic differentiation conditions.
In addition, we proved that AT-SCs after lentiviral-mediated gene transfer provide robust transgene expression. In particular, we generated LVs expressing enhanced green fluorescent protein (E-GFP), human α1-antitrypsin (hAAT), and firefly luciferase (fluc) under the ubiquitous human phosphoglycerate kinase (PGK) promoter. Upon lentiviral-mediated gene transfer at multiplicity of infection (MOI) of 10, we achieve up to 95% transduction, as assessed by E-GFP expression determined by FACS and fluorescent microscopy analysis (Fig. 2B). Moreover, after LV-mediated gene transfer of hAAT into murine AT-SCs, overexpression and secretion of hAAT was achieved (Fig. 2C).

Reporter expression after lentiviral-mediated gene transfer into AT-SCs. Before transplantation, AT-SCs were genetically modified by lentiviral-mediated gene transfer. In particular, we used lentiviral vectors expressing enhanced green fluorescent protein (E-GFP), human a1-antitrypsin (hAAT), or firefly luciferase (fluc) under the ubiquitous human phosphoglycerate kinase (PGK) promoter. Bright field (A) and UV light (B) microphotographs of AT-SCs transduced with LV-E-GFP at multiplicity of infection (MOI) 10, 72 h after gene transfer. FACS analysis of transduced cells indicated up to 95% GFP-positive cells. Scale bar: 100 μm. (C) Secretion of hAAT in the culture medium of LV-hAAT-transduced AT-SCs under the same conditions. Experiments were performed three times, each time in duplicate. Data are expressed as means ± SEM as indicated by error bars. (D) Ex vivo bioluminescence imaging (BLI) analysis of AT-SCs after fluc gene transfer. No signal was detected in mock-transduced cells (not shown). Representative example showing a portion of a tissue culture 96-well plate; experiment was performed in quadruplicate.
Taken together, these data suggest that murine AT-SCs derived from inguinal fat pads have hepatogenic transdifferentiation properties in vitro in the presence of selective inductive media, in accordance with previous results (37).
Transplantation of Ex Vivo LV-Transduced AT-SCs in a Murine Model of Liver Injury
We next evaluated whether AT-SCs after minimal in vitro expansion and LV gene manipulation have the ability to engraft in an animal model of hepatic liver injury.
In order to follow the fate of transplanted cells, AT-SCs were transduced ex vivo with LV expressing luciferase under the control of the ubiquitous PGK promoter. Luciferase activity in transduced cells was determined by enzymatic luciferase assay (data not shown) and by bioluminescence imaging (Fig. 2D). A positive signal was detected with as low as 5.0 × 103 cells 2 days after LV-fluc-mediated gene transfer (Fig. 2D), and it was maintained at a similar level after up to 8 weeks of culture (data not shown).
Consistent lines of evidences have demonstrated that transplanted cell engraftment can be improved by different methods of recipient liver preconditioning [reviewed in (50)]. We induced hepatic damage in Swiss CD1 mice by intraperitoneal injection of CCl4 a day before cell transplantation (27). Intrasplenic transplantation is a well-established route to deliver hepatocytes to the liver (29). We proved that this route is also the optimal route for hepatic delivery of AT-SCs. Conversely, systemic administration by intratail vein injection or intraperitoneal delivery resulted in cell translocation into nonhepatic sites as assessed by in vivo imaging (Fig. 3), a problem possibly correlated with the size of the injected cells. In particular, in order to determine biodistribution of cells administered via different routes of administration, we performed in vivo imaging within 1 h after administration of identical doses of luciferase-expressing AT-SCs into mice after CCl4-mediated liver injury. The bioluminescence image was overlaid on a light picture of the animal as reference for anatomical localization; moreover, BLI was performed after animals' sacrifice and dissection and on isolated organs (Fig. 3). After intrasplenic delivery, cells were localized within the spleen and the liver (Fig. 3D, E, F). After systemic administration by intratail vein injection, all BLI signals were localized in lungs, indicating cell trapping within lung capillaries (Fig. 3G, H, I) (35). After intraperitoneal administration, AT-SCs were retained within the peritoneal cavity and were not detectable in heart, lungs, spleen, or liver (Fig. 3J, K, L).

Biodistribution after AT-SCs transplant. Swiss CD1 mice were treated with intraperitoneal injection of carbon tetrachloride (CCl4; 1 ml/kg) a day before transplantation of AT-SCs (2.5 × 105 cells/mouse in 100 μl of saline solution). In vivo bioluminescence imaging was performed within 1 h after transplant. Mice were divided in four groups of four animals each. One group of animals received mock-transduced cells by intrasplenic administration (A, B, C). The other groups received AT-SCs transduced with LV-fluc by intrasplenic (D, E, F), intratail vein (G, H, I), or intraperitoneal administration (J, K, L). (A, D, G, J) In vivo imaging performed on a representative mouse of each group. (B, E, H, K) Same animals after sacrifice and dissection. (C, F, I, L) BLI performed on a portion of dissected organs (h, heart; lu, lungs; li, liver; s, spleen). Scale bars: 1 cm.
In subsequent studies, Swiss CD1 mice were treated intrasplenically with AT-SCs expressing firefly luciferase 24 h after CCl4-induced hepatic injury. In vivo imaging was performed at different time points beginning at day 1 after transplant (Fig. 4). The signal decreased progressively and, by approximately 3 weeks, was no longer detectable in the spleen by in vivo BLI in all mice (n = 8), suggesting that AT-SCs do not engraft into the spleen. Conversely, luminescence was detectable in the liver for up to 2 months, indicating persistence of administered cells at the site of hepatic injury.

Cell tracking and kinetic of engraftment by bioluminescence imaging of AT-SCs expressing firefly luciferase. Swiss CD1 mice were injected intraperitoneally with CCl4 (1 ml/kg) a day before intrasplenic transplantation of AT-SCs transduced with LV-fluc (2.5 × 105 cells/mouse). Longitudinal bioluminescence imaging of AT-SCs expressing luciferase in a representative animal (n = 8) and quantification of the signal intensity (expressed in photons/s/cm2/sr). All control animals administered with non-luciferase-expressing cells (n = 6) or with PBS only (n = 2) were negative for BLI signal at all time points (data not shown). Scale bar: 1 cm.
The same animals were monitored at different time points after cell administration to longitudinally track and quantify cell engraftment. Luminescence decreased dramatically during the first few days after transplantation, possibly due to the rigors of the transplanted cells' new microenvironment. Up to 90% of the BLI signal assessed in liver and spleen at 1 day was lost within the first 2 weeks after transplant. Nonetheless, engraftment of a small proportion of cells was maintained up to 2 months after administration in more than 80% of the analyzed animals. The signal strength for each locus of engraftment was comparable throughout the analyzed period (Fig. 4). Bioluminescence at 2 months was approximately 1% of the signal assessed 1 day after transplant.
Upon sacrifice, livers were dissected and analyzed by the IVIS® Lumina imaging system and by immunohistochemistry (Fig. 5). The presence of luciferase-positive cells was detected by BLI in several regions of the liver of animals transplanted with luciferase-expressing AT-SCs (Fig. 5E) and not in mock-treated controls (Fig. 5A). Immunohistochemical analysis on liver sections of transplanted mice confirmed the presence of luciferase positive cells whose morphology resembles that of hepatic cells (Fig. 5H). No fluorescent staining could be observed with the anti-luc antibody around the regenerating region in the uninjected controls (Fig. 5D).
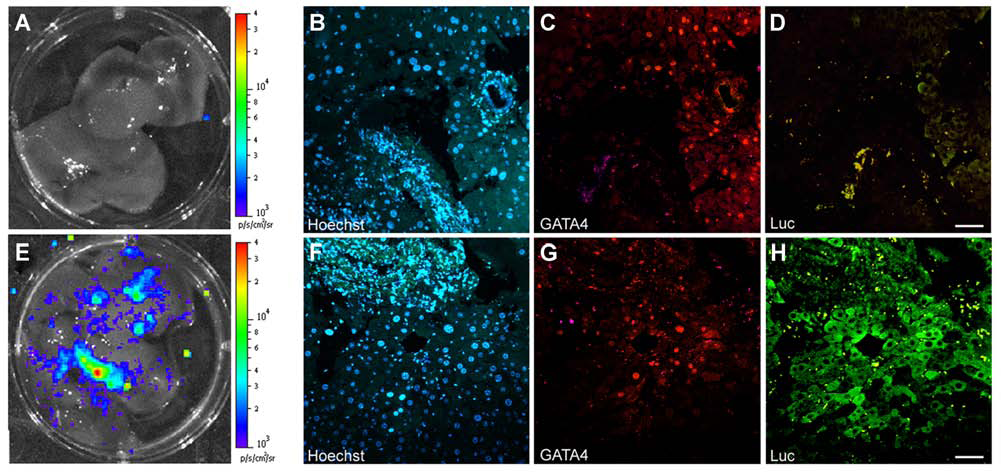
Engraftment of AT-SCs expressing firefly luciferase at the site of liver injury at 2 weeks postadministration. (A, B, C, D) An animal subjected to CCl4-mediated injury but without cell transplantation. (E, F, G, H) A mouse receiving CCl4-mediated injury and intrasplenic transplantation of luciferase expressing AT-SCs. Ex vivo imaging of dissected liver from a representative mouse of each group (A, E), denoting the presence of luciferase-positive cells in transplanted animals only. Images show dissected organs placed in a well of a six-well tissue culture plate. (B) Immunohistochemistry on consecutive liver sections using antibodies against either GATA4 (red) or firefly luciferase (green). Nuclei are visualized by Hoechst dye (blue). Scale bars: 100 μm.
GATA transcription factors are a conserved family of zinc finger-containing proteins that participate in the specification and differentiation of multiple cell types during development. In particular, GATA4 plays a pivotal role in controlling the earliest stages of hepatic development (40). GATA4 is highly expressed in hepatocytes and endothelial cells during early development, whereas GATA4 expression in the adult liver is low and restricted to epithelial cells surrounding the biliary ducts (48). However, GATA4 is reexpressed in regenerating liver (25). LV-transduced, luciferase-expressing AT-SCs (Fig. 5H) overlapped with GATA4 expression as assessed by immunohistochemical analysis on liver sections (Fig. 5G). This indicates that transplanted cells predominantly colocalize with regions of active hepatic regeneration following CCl4-induced injury. Moreover, GATA expression observed in luciferase-positive cells indicates that transplanted cells adopt a hepatic-like phenotype and participate in tissue regeneration.
In order to determine whether the rate of engraftment can support prolonged expression and secretion into the bloodstream of a transgene with potential therapeutic index, we genetically modified AT-SCs to overexpress human AAT. AT-SCs transduced either with a mock LV or an hAAT-expressing LV vector were administered into the spleen, as described above. Blood samples were obtained at different time points after transplant. Plasma levels of human AAT were measured as a correlate of AT-SC engraftment after hepatic injury (14). Levels of hAAT were undetectable in all animals before transplant and in animals undergoing transplantation with mock-transduced cells. On the other hand, hAAT was detectable in mice receiving intrasplenic administration of AT-SCs genetically modified with LV-hAAT up to 7 weeks after transplantation (Fig. 6).
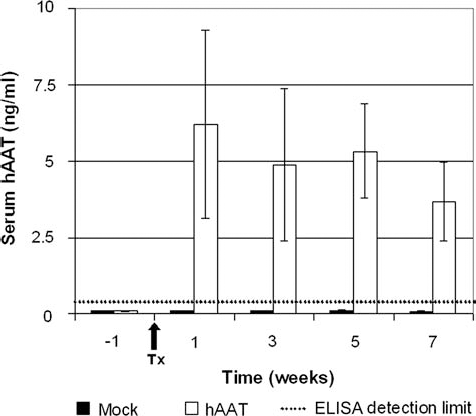
Serum hAAT levels in mice after intrasplenic transplant of AT-SCs overexpressing hAAT. Swiss CD1 mice were treated with intraperitoneal injection of CCl4 (1 ml/kg) a day before intrasplenic transplantation of AT-SCs transduced either with LV-E-GFP (AT-SCs Mock) or LV-hAAT (AT-SCs hAAT) (2.5 × 105 cells in 100 μl saline solution per mouse). Blood was drawn at multiple time points, and serum hAAT levels were quantified by ELISA. The dotted line indicates ELISA limit of detection (0.4 ng/ml). Tx indicates the day of transplantation. Results are means ± SEM of six animals per group. All p values between AT-SCs hAAT-treated animals compared to AT-SCs mock mice were p < 0.005.
These data provide evidence that the amount of engrafted AT-SCs is sufficient to secrete detectable levels of protein for almost 2 months.
Activation of AFP Promoter in Transplanted Cells Reisolated From Injected Livers
AFP is a suitable marker for hepatic differentiation since it is expressed during fetal liver development and regeneration but not in healthy adult tissue (17). AT-SCs expressing luciferase were transduced with LVs expressing GFP under the control of the specific AFP promoter (LV-AFPPr. EGFP). Cells were then transplanted as described before, and animals were sacrificed 10 days later. Site of transplanted cell engraftment were detected by BLI; corresponding regions were dissected, disaggregated by collagenase digestion, and harvested for cell culture. GPF-positive cells were detected by fluorescent microscopy in cultures of cells isolated from animals transplanted with LV-AFPPr. EGFP-transduced AT-SCs, indicating an activation of the fetal liver-specific AFP enhancer/promoter at the site of liver engraftment (Fig. 7).

In vivo differentiation of AT-SCs tagged with an AFP promoter-driven reporter. AT-SCs were transduced ex vivo with lentiviral vectors driving E-GFP expression under the control of the liver-specific α-fetoprotein promoter (AFPPr.EGFP). The hAFP promoter is inactive in undifferentiated AT-SCs (see Fig. 1). Cells were then transplanted by intrasplenic injection in syngeneic animals. Ten days later, livers were dissected, hepatic cells were isolated and cultured, and GFP-positive cells were visualized under a fluorescent microscopy. Scale bar: 100 μm.
These data suggest that physiological environmental cues present at the site of engraftment in the injured liver are sufficient to promote GFP expression in engrafted cells under the control of the AFP promoter, which indicates differentiation into hepatocyte-like cells.
Discussion
Liver stem or precursor cells are emerging as promising therapeutic tools (23,42). Recently, a Crigler–Najjar patient was treated by infusion of hepatic progenitor cells isolated from fetal liver (15); moreover, several patients affected by liver cirrhosis have been treated by autologous bone marrow cell transplant (32). Additional clinical trials using adipose tissue-derived stromal cells aiming at liver regeneration have been proposed (22). The treatment of liver diseases with AT-SC-derived hepatic cells may have considerable advantages over the use of cells of different origin (46). Adipose tissue can be obtained with a simple, minimally invasive, and repeatable procedure. A clinically relevant number of cells can be harvested, reducing the need for ex vivo cell expansion in GMP conditions, and further avoiding genomic instability and the difficulty in maintaining undifferentiated pluripotent cells. Nevertheless, for the treatment of liver genetic disorders, these cells would be obtained from a different donor, necessitating immunosuppressive treatment and its associated morbidity.
Ex vivo gene delivery into progenitor cells followed by autologous transplant may provide for the treatment of different metabolic disorders currently lacking adequate therapeutic options. Nonetheless, many issues are unclear, including the specific contribution of transplanted cells to physiologic tissue regeneration and the biodistribution of cells upon administration.
Recently Aurich et al. (1) described that in vitro hepatogenic predifferentiation of human AT-SCs facilitates functional hepatic integration in vivo. Our data show that murine AT-SCs are susceptible to ex vivo hepatic induction similarly to human cells. However, all inductive media formulations used for hepatogenic in vitro predifferentiation (1,2,20) contain nonphysiological levels of cytokines, growth factors, and hormones and therefore raise serious safety concerns over the suitability of this method for clinical trials. Moreover, most results have been produced on AT-SCs cultured in medium supplemented with FBS, while data on cells cultured with clinically relevant human serum derivatives or serum-free conditions are incomplete (21). Finally, the prolonged in vitro culture, required for efficient differentiation, is associated with decreased proliferation, increased cell size, and chromosomal instabilities (47). Due to these limitations, we decided for the subsequent transplantation study to use cells after minimal ex vivo manipulation in noninductive basal medium. In this case, support for the engraftment, differentiation, and prolonged survival of transplanted cells is provided by environmental cues in the host liver. Our data show that endogenous signals are sufficient for AT-SCs to acquire hepatocyte-like characteristics, such as hepatocyte morphology, concomitant GATA4 expression, and the activation of the AFP promoter.
Different animal models of hepatic injury have been investigated for studying liver repopulation (11). In most of these models, transplanted cells have a selective advantage over resident hepatocytes. For instance, Aurich et al. (1) used 30% partial hepatectomy and monocrotaline treatment for inhibition of proliferation of resident liver cells, allowing transplanted cells to repopulate the organ (49). However, monocrotaline treatment is not clinically translatable. In our study, we assessed AT-SCs engraftment in the liver in the absence of any selective advantage.
Recently, near infrared fluorescence has been used for cell tracking after HcTx in a rat experimental model (16). BLI involves the detection of photons from cells expressing a luciferase enzyme. This procedure offers various advantages over alternative fluorescence-based molecular imaging techniques, including a high signal to background ratio allowing for more sensitive, longitudinal, and quantitative analysis (5,31) also into the liver (4). The intensity of the signal detected by BLI can be precisely quantified and correlates with the presence of luciferase-expressing cells and therefore with effective cell engraftment after administration. Since luciferin metabolism requires ATP to generate light, only living cells expressing luciferase are able to produce a signal (31). The possibility to perform repeated analysis on the same animal at different time points allowed us to follow the fate of AT-SCs after transplantation into CCl4-injured mice. In particular, we demonstrated that AT-SCs are capable of migrating through the spleen, engrafting in the liver into regenerating sites, and persisting in the hepatic parenchyma for up to 2 months. This is, to our knowledge, the first description of the use of BLI to monitor an experimental approach of cell therapy for liver disorders using adipose tissue-derived stromal cells.
In this study, we concomitantly evaluated a cell therapy and a cell-mediated gene therapy approach as possible treatments for hepatic disorders. We show that AT-SCs are prone to ex vivo genetic manipulation by lentiviral-mediated gene transfer without affecting their differentiation capacity. Therefore, such cells could be used either for allogeneic transplantation or for autologous transplant after ex vivo genetic modification to promote expression of a therapeutic transgene, considerably reducing the risks associated with systemic viral exposure. The possibility to manipulate and transplant autologous cells eliminates the need for lifelong immunosuppression. Of note, it has been demonstrated that some immunosuppression treatments, applied towards human cell transplantation in rodents and for heterologous hepatocyte transplantation, have a detrimental effect on the engraftment and proliferation of implanted hepatocytes (41).
Hepatocytes transplanted by the intrasplenic route reconstitute 1–5% of the host hepatocyte mass (29). Hepatic reconstitution to this extent may be sufficiently therapeutic for metabolic disorders requiring proteins in small amounts, such as hemophilia. After transplantation of hAAT-expressing AT-SCs, we observed clearly detectable but subphysiological levels of circulating hAAT. In such cases, strategies to increase transgene expression and/or cell engraftment are needed. Nevertheless, we provide proof of concept for secretion of a soluble factor by genetically engineered, engrafted AT-SCs.
In conclusion, our study shows that AT-SC-based cell therapy may represent an attractive clinical option for the treatment of both degenerative and metabolic liver diseases. AT-SCs can acquire hepatic-like characteristics both in vitro and in vivo, and genetically modified AT-SCs can engraft into the regenerating liver, providing durable transgene expression in the absence of any selective advantage. This validates the prospective use of AT-SCs as a platform for autologous cell-mediated gene delivery in hepatic regeneration.
Footnotes
Acknowledgments
We thank Isabella Manni and Simona Artuso (Istituto Regina Elena IFO-CRS) for assistance for BLI and Viraj P. Mane (University of Maryland, College Park) for critical review of the manuscript. Financial support by the Crigler–Najjar Italia-Associazione Malati Iperbilirubinemici (CIAMI Onlus) and by the Dino Bianchi-Edison Award 2009 donation is gratefully acknowledged. The authors declare no conflicts of interest.