
Abstract
Development of β-cells from human embryonic stem cells (hESCs) could compensate for the shortage of islet donors required for diabetes therapy. Although pancreatic progenitors have been derived from hESCs using various protocols, no fully functional β-cells could be generated in vitro. We evaluated the in vivo growth and differentiation of PDX1+ pancreatic endoderm cells obtained from hESCs. Here we show site-specific survival and differentiation when comparing cells grafted in the epididymal fat pad or the subcutaneous space of NOD/SCID mice after 12 weeks follow-up. Subcutaneous grafts persisted and expressed PDX1 at all time points analyzed, showed PDX1 and NKX6.1 coexpression after 6 weeks, and contained NGN3+ cells after 12 weeks. These findings suggest that further specification along the pancreatic lineage occured at the subcutaneous site. In sharp contrast, in the fat pad grafts only a minority of PDX1+ cells remained after 2 weeks, and no further pancreatic differentiation was observed later on. In addition, contaminating mesenchymal cells present in the implants further developed into cartilage tissue after 6 weeks implantation in the fat pad, but not in the subcutaneous space. These findings indicate that the in vivo microenvironment plays a critical role in the further differentiation of transplanted pancreatic endoderm cells.
Keywords
Introduction
Insulin-dependent diabetes is currently an incurable chronic disease. As an alternative to life-long exogenous insulin injections and their complications, pancreas or islet cell transplantation represents a promising therapy (23). However, the shortage of pancreas donors makes transplantation therapy available to only a minority of patients. This problem may be overcome by transplanting β-cells derived from adult or embryonic stem cells (2,19,20,27). Human embryonic stem cells (hESCs) that are characterized by their ability of unlimited expansion and differentiation to every type of tissue have been considered as a promising resource of cell therapy for curing diabetes. Since the first demonstration of low-frequency spontaneous pancreatic differentiation from hESCs (1), a number of protocols have been developed to generate β-like cells from these cells. In 2006, D'Amour et al. (5) developed a five-stage protocol to generate insulin-producing cells from hESCs by mimicking the main events that govern (murine) pancreas development in vivo. Although a small proportion of insulin-positive cells could be obtained by this protocol and by similar protocols, these cells showed poor (9,21) or absent (5,13) insulin secretory response to glucose. In vivo transplantation of pancreatic endocrine progenitor cells derived from hESCs allowed their further differentiation into functional glucose-responsive insulin-expressing cells capable of reverting hyperglycemia in diabetic mice (10,12,24). These findings suggest that currently unknown molecular signals, play a crucial role in the differentiation of hESC-derived pancreatic endocrine cells in vivo. The nature of the signals favoring the differentiation process in vivo remains to be identified before fully functional β-cells can be generated in culture. Despite the promising data obtained by D'Amour et al. (5) in vitro and by Kroon et al. (12) in vivo, similar results could not be replicated by other research groups with different hESC lines in vitro (3,17,22) or a different animal model in vivo (15). These limitations imply potential difficulties in implementing this method into practice. To tackle this problem, we recently developed an efficient and widely applicable protocol (17) to differentiate hESC lines into Pancreatic and duodenal homeobox 1 (PDX1)-positive pancreatic endoderm (PPP).
In the present study, we tested whether the PPP cells derived from hESCs in our lab are really committed to the pancreatic lineage and whether they can further mature in vivo. Transplantation experiments were performed in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice to assess the survival, proliferation, and differentiation of PPP cells. We found that PPP cells have the potential to further differentiate into fully specified pancreatic progenitors and endocrine progenitors in vivo. Unexpectedly, we found a transplantation site-specific survival and differentiation ability of the grafted cells.
Material and Methods
Maintenance and Differentiation of hESCs
Human ES cell line VUB07 was generated and characterized at our institute (14). It is registered at www.hescreg.eu (European Human Embryonic Stem Cell Registry). Undifferentiated cells were maintained on mitomycin C-treated primary mouse embryonic fibroblasts (Millipore, Billerica, MA) and manually passaged every 5–7 days (17).
Differentiation was carried out in three stages as described previously (17). Briefly, hESCs were washed three times with PBS before initiating stage 1 in Roswell Park Memorial Institute medium (RPMI-1640; Invitrogen, Carlsbad, CA) supplemented with 100 ng/ml Activin A (R&D Systems, Minneapolis, MN) and 25 ng/ml wingless-type MMTV integration site family, member 3A (Wnt3a; R&D Systems) for 1 day, followed by Activin A and 0.2% fetal bovine serum (FBS; PAA Laboratories, Pasching, Austria) for another 2 days in order to induce definitive endoderm (DE). At stage 2, 100 ng/ml Noggin (R&D Systems), 250 nM 3-Keto-N-(aminoethyl-aminocaproyl-dihydrocinnamoyl) cyclopamine (KAAD-cyclopamine; Cyclo, Calbiochem, Darmstadt, Germany), and 2 μM retinoic acid (RA, Sigma-Aldrich, St. Louis, MO) were added in RPMI-1640 + 2% FBS for 4 days and in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) plus 1% B27 (Invitrogen) for another 4 days to block the hepatic differentiation and induce pancreatic differentiation. At stage 3, cells were cultured in DMEM + 1% B27 containing 50 ng/ml fibroblast growth factor (FGF10; R&D Systems), 1 μM γ-secretase inhibitor Compound-E (CpdE, Calbiochem), and 50 ng/ml exendin-4 (Ex4, Sigma-Aldrich) for 4 days to obtain the PPP cells (Table 1). Media were renewed every other day.
Schematic Overview of the Directed Differentiation of Human Embryonic Stem Cells (hESCs) Into PPP Cells

Cells were cultured on inactivated mouse embryonic fibroblasts and differentiated at 80% confluence. Media and factors were refreshed every other day and samples harvested at determined time points for analysis or transplantation. DE, definitive endoderm stage; HBPI, hepatocyte blockade and pancreas induction stage; PPP, Pancreatic and duodenal homeobox 1 (PDX1)-positive pancreatic endoderm stage; RPMI, Roswell Park Memorial Institute medium; DMEM, Dulbecco's modified Eagles Medium; ActA, Activin A; Wnt3a, wingless-type MMTV integration site family, member 3A; FBS, fetal bovine serum; Cyclo, KAAD-cyclopamine; RA, retinoic acid; FGF10, fibroblast growth factor 10; CpdE, γ-secretase inhibitor compound-E;Ex4, exendin-4.
Transplantation of PPP Cells
Male immune-deficient NOD/SCID mice (8–10 weeks old; Charles River Laboratories, L'Arbresle, France) were housed according to the guidelines of the Belgian Regulations for Animal Care. A total of 21 mice were implanted from 12 independent PPP cell preparations. Prior to transplantation, PPP stage cells were washed with PBS and dissociated with TrypZean (Sigma-Aldrich) into small aggregates. About 1–2.5 million PPP cells were implanted into either the epididymal fat pad or the dorsal subcutaneous space of NOD/SCID mice. As cell carriers, we initially compared fibrin glue (Tissucol, Baxter, Deerfield, IL) and Gelfoam sponge overlayed with Matrigel (BD Biosciences, San Jose, CA) but noticed no differences in the graft characteristics. Gelfoam sponges were preferred for the ease of operation. Mice were euthanized at 2, 6, 9, and 12 weeks postengraftment, and grafts were retrieved for analysis.
Quantitative Real-Time PCR
Total RNA was extracted at different stages (hESCs, DE, and PPP) prior to transplantation using the GenElute mammalian total RNA kit (Sigma-Aldrich) and the concentration and quality assessed on a ND-1,000 spectrophotometer (Thermo-Scientific, Waltham, MA). RNA (500–1,000 ng) was reverse-transcribed using the Verso cDNA kit (Thermo-Scientific). Finally, a cDNA aliquot corresponding to 28 ng RNA equivalent was added to 250 nM primers in SYBR Green master mix (Thermo-Scientific) and loaded onto a 96-well plate compatible with the ABI-7900HT real-time PCR machine (Applied Biosystems, Foster City, CA). The sequences of the primers used are listed in Table 2. Relative quantification of gene expression was calculated by the ddCt method and normalized to endogenous control TATA-binding protein (TBP). Data are presented as fold change versus undifferentiated hESCs.
List and Sequences of Primers Used in Quantitative PCR

The primers were designed preferentially in an intron-spanning region with an amplicon size less than 200 bp. TBP, TATA-binding protein; FOXA2, forkhead protein A2; SOX17, sex determining region Y box 17; INS, insulin; NKX6.1, NK6 homeobox 1; NGN3, neurogenin 3; CK19, cytokeratin 19; P48, pancreas specific transcription factor 1a (PTF1a).
Immunofluorescence
Cultured cells were washed with PBS and fixed in the four-well plates at DE and PPP stages with 4% formaldehyde for 15 min at room temperature. Cells were permeabilized in cold methanol (-20°C) for 20 min and blocked for 30 min in 2% normal donkey serum. Primary antibodies were diluted and incubated with cells at 4°C overnight. Secondary antibodies were diluted and incubated at room temperature for 45 min. Nuclei were stained with Hoechst. The following primary antibodies and dilutions were used: goat anti-forkhead protein A2 (FOXA2), 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA; SC-9187); mouse anti-sex determining region Y box 17 (SOX17), 1:100 (R&D Systems; MAB1924); goat anti-PDX1, 1:100 (R&D Systems; AF2419); mouse anti-NK6 homeobox 1 (NKX6.1), 1:250 (Developmental Studies Hybridoma Bank, Iowa City, IA; F55A10); sheep anti-neurogenin 3 (NGN3), 1:100 (R&D Systems; AF3444); rabbit anti-SOX9, 1:500 (Millipore; AB5535); mouse anti-cytokeratin-19 (CK19), 1:25 (DAKO, Glostrup, Denmark; M0888); rabbit anti-human a fetoprotein, 1:200 (AFP; Dako; A0008); mouse anti-human mitochondria, 1:50 (HU; Millipore; MAB1273); mouse anti-E-cadherin, 1:50 (BD Biosciences; 610182); rabbit anti-Ki67, 1:5,000 (Novocastra Laboratories, Newcastle upon Tyne, UK; NCL-Ki67p); mouse anti-human vimentin, 1:200 (DAKO; M0725); guinea pig anti-insulin, 1:100 (gift from Prof C. Van Schravendijk, VUB, Belgium). Secondary antibodies were Alexa Fluor 488- and 555-conjugated donkey anti-goat and mouse, 1:500 (Invitrogen); fluorescein isothiocyanate (FITC)-labeled donkey anti-rabbit, 1:200; tetramethyl rhodamine isothiocyanate (TRITC)-labeled donkey anti-mouse, 1:100; TRITC-labeled donkey anti-sheep, 1:100; cyanine 3 (CY3)-labeled donkey anti-guinea pig, 1:200 (all from Jackson ImmunoResearch Laboratories, West Grove, PA). Negative controls were performed by ommission of primary antibodies during the same staining procedure.
Tissues harvested from the grafts at determined time points were fixed in 4% formaldehyde for 4 h at room temperature and embedded in paraffin. The specimens were then serially sectioned all through and selected slides (one every 10 slides) were stained with hematoxylin and eosin (H&E) to detect the grafted cells. Deparaffinization was performed by heating the slides at 60°C for 1 h followed by immersion in toluol for 10 min. The slides were further rehydrated in decreasing percentages of ethanol solutions and subjected to immunostaining following the same protocol used for cultured cells. All stained samples were examined for immunofluorescence under a Nikon Eclipse TE-2000 immunofluorescence microscope, and the pictures were captured and analyzed using the NIS Elements software (Nikon, Tokyo, Japan).
Statistical Analysis
All experiments in vitro were performed at least three times independently. Data were analyzed by one-way ANOVA followed by Bonferroni's multiple comparison test (GraphPad Prism 5, GraphPad Software, Inc., La Jolla, CA) and expressed as mean ± standard error of the mean (SEM). For the transplantation studies, we compared the outcomes of grafted cells in subcutaneous implantation vs. epididymal fat pad implantation using the Fisher exact test. The differences observed were considered statistically significant at the 5% level and were displayed on the figures as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Characteristics of the Grafted Cells Preimplantation
We have recently established a method for hESC differentiation into pancreatic endoderm (Table 1) (17). To test the specificity of antibodies, we first examined the undifferentiated hESCs with several markers that will be later used for evaluating the phenotype of differentiated cells (Fig. 1A). The human origin of hESCs was confirmed as indicated by the positive staining for human-specific mitochondria. They were also positive for E-cadherin and negative for the mesenchymal marker vimentin, DE markers FOXA2 and SOX17, and pancreatic endoderm marker PDX1. We tested that the cells in a first stage (3 days) differentiated into DE by showing increased expression of DE markers FOXA2 and SOX17 with respect to undifferentiated hESCs (Fig. 1B). In immunocytochemistry, 80% of cells stained positive for FOXA2 and SOX17 (Fig. 1C), indicating efficient DE induction in culture. After DE induction, bone morphogenetic protein (BMP) and Hedgehog pathways were blocked to exclude liver and intestinal lineages, while RA was added to induce expression of PDX1 (stage 2). Finally, FGF10, CpdE, and Ex4 were combined with the intention of inducing pancreatic progenitors and endocrine progenitors (stage 3). At the end of the differentiation protocol, pancreatic endoderm (PPP stage) was obtained with significantly increased expression of pancreatic endoderm markers PDX1, SOX9, and CK19 and stable FOXA2 expression (p < 0.01) (Figs. 1B, 2A). At this stage, expression of other pancreatic progenitor markers NKX6.1, pancreas transcription factor 1 subunit α (PTF1α/P48) or endocrine markers NGN3 and the β-cell marker Insulin (INS) were marginally increased (Fig. 2A). To further confirm the results at the protein level, pancreatic endoderm and progenitor markers were immunostained at the end of PPP stage (Fig. 2B). Those PPP cells expressed CK19, FOXA2, PDX1, and SOX9 and were negative for the hepatic marker α-fetoprotein (AFP). We observed that 60–80% of cells were stained with PDX1 and SOX9 but only a few of them coexpressed NKX6.1 (Fig. 2B), although some NKX6.1+ cells were detected in PDX1-negative area as previously described (17). The contaminating 20–40% nonpancreatic cells appeared to be mainly of mesodermal origin as indicated by expression of the mesenchymal marker vimentin (Fig. 2C). Our findings confirm that a high proportion of pancreatic endoderm cells (expressing PDX1, FOXA2, SOX9, and CK19) could be derived from hESCs, but these cells are still early in the pancreatic differentiation program as indicated by low expression of NKX6.1, PTF1α/P48, and NGN3.

Differentiation of human embryonic stem cells (hESCs) into definitive endoderm (DE) cells in vitro. (A) A panel of antibodies against markers to be analyzed at later differentiated stages was evaluated on undifferentiated hESCs for quality control. All the hESCs colonies were positive for human-specific mitochondria protein (HU) and E-cadherin (E-Cad). Only few, if any, of them expressed vimentin, forkhead box protein A2 (FOXA2), sex determining region Y box 17 (SOX17), and pancreatic and duodenal homeobox 1 (PDX1). (B) Transcripts of the DE markers FOXA2 and SOX17 were induced during the 3 initial days of culture. PPP, PDX1-positive pancreatic endoderm. (C) At the DE stage, the majority of cells coexpressed FOXA2 and SOX17 proteins. Scale bar: 50 μm.

Differentiation of hESCs into PPP cells in vitro. (A) The pancreatic progenitor markers were induced during the 15 days of culture. The expression of PDX1, SOX9, and cytokeratin 19 (CK19) all significantly increased, whereas the expression of NK6 homeobox 1 (NKX6.1), pancreas transcription factor 1 subunit α (PTF1α/P48), neurogenin 3 (NGN3), and insulin (INS) were just marginally changed. (B) At the PPP stage, around 70% cells expressed PDX1, SOX9, CK19, and FOXA2, few of them expressed NKX6.1. They were negative for the hepatic marker α-fetoprotein (AFP). (C) Beside PDX1+ and SOX9+ pancreatic endoderm cells, a group of vimentin+ (20–40%) cells were detected at the PPP stages. Scale bar: 50 μm.
Histological Analysis of the PPP Cells After Transplantation
To assess the differentiation potential of the in vitro hESC-derived pancreatic endoderm, we grafted PPP stage cells in the epididymal fat pad (n = 16) and in the subcutaneous space (n = 18) of NOD/SCID mice. We examined graft histology at 2, 6, 9, and 12 weeks after implantation in both sites. A human-specific anti-mitochondria antibody was used to identify the grafted cells, and E-cadherin antibody was used to distinguish epithelial cells.
Two Weeks Postimplantation
At 2 weeks after implantation, five grafts from the epididymal fat pad were analyzed. Only two of them contained a small group of PDX1+ cells, the majority of which were negative for the proliferation marker Ki67 (Fig. 3A). Most of the Ki67+ cells appeared in the E-cadherin negative area corresponding to mesenchymal cells. The other three grafts showed either no surviving cells or mainly mesenchymal-like cells of human origin as indicated by their expression of human vimentin (Fig. 3B). These observations suggest that PDX1+ cells did not survive well and were replaced by mesenchymal cells in the fat pad grafts. In sharp contrast, all five grafts implanted in the subcutaneous space displayed duct-like or cystic epithelial structures at 2 weeks. Most of the epithelial cells coexpressed PDX1, SOX9, and CK19 (Fig. 3C), and several PDX1+ cells also stained positive for Ki67 (Fig. 3D). Neither NKX6.1-expressing cells nor tumor formation could be detected in the grafts implanted in both sites at this time point. These observations suggest that the subcutaneous space is a better site to accomodate PPP cells than the fat pad.

Immunofluorescence analysis of fat pad (A, B) and subcutaneous (C, D) grafts at 2 weeks posttransplantation. (A) In the fat pad, grafted cells were detected 2 weeks after transplantation by staining for human-specific anti-mitochondria antibody. A small group of PDX1+ epithelial cells was observed. E-cadherin-positive epithelium corresponded to the HU-positive area, and the PDX1+ cells appear negative for Ki67 (in the trapezoid). (B) Beside PDX1+ cells, mesenchymal cells of human origin were also detected as evidenced by staining for human-specific vimentin antibody. (C) In subcutaneous space, 2 weeks grafted cells expressed PDX1, SOX9, and CK19. (D) All the PDX1+ cells were positive for human-specific antibody, and most of them coexpressed Ki67 (outlined boxes). Scale bar: 50 μm.
Six to Twelve Weeks Postimplantation
We then analyzed the grafts after 6, 9, or 12 weeks implantation in the epididymal fat pad (seven grafts at 6 weeks, three at 9 weeks, and one at 12 weeks) or the subcutaneous space (six grafts at 6 weeks, five at 9 weeks, and two at 12 weeks). Overall, teratoma formation was detected in three of 24 grafts (two in the fat pad, one in the subcutaneous space), whereas grafts without surviving cells were identified twice (both in the subcutaneous space). In none of the epididymal fat pad grafts could we find significant numbers of PDX1+ cells nor PDX1-NKX6.1 double-positive cells. Unexpectedly, a large mass of chondrocyte-like cells were noticed as characterized by cartilage morphology and by the expression of SOX9, a transcription factor involved in chondrocytic differentiation (Fig. 4A). Coexpression of human-specific mitochondrial antigen indicated that these chondrocytes were derived from the grafted human cells (Fig. 4B).

Immunofluorescence analysis of fat pad (A, B) and subcutaneous (C, D) grafts at later time point (more than 6 weeks) posttransplantation. (A) After 6 weeks engraftment, the PDX1+ cells vanished gradually and large areas of chondrocytes were detected recognized by their morphology in hematoxylin and eosin (H&E)-stained sections and the expression of SOX9. (B) Coexpression of human-specific antigen indicated that those chondrocytes were of human origin (outlined by dotted line). (C) Some of the PDX1+ cells coexpressed NKX6.1 and with potential to proliferate as indicated by expression of Ki67. (D) The PDX1 and NKX6.1 costained cells always formed small clusters (c) and were located near by a PDX1+ duct-like structure (d). Scale bar: 50 μm.
In the subcutaneous environment, all the well-developed grafts (n = 10) contained large areas of PDX1+ cell clusters organized in duct-like or cystic structures. No chondrocyte-like cells were found in the grafts at this site. In 50% (5/10) of the subcutaneous grafts analyzed between 6 and 12 weeks, PDX1+ cells coexpressed NKX6.1, whereas no coexpression was noted at 2 weeks. In addition, these cells showed proliferation ability (Fig. 4C). The PDX1-NKX6.1 double-positive cells always formed small clusters along PDX1+ duct-like structures (Fig. 4D). At the latest time point examined (12 weeks), we also detected some NGN3+ endocrine progenitors (n = 1) as well as insulin+ endocrine cells that coexpressed the transcription factor NKX6.1, indicating further differentiation along the pancreatic endocrine progenitor fate (Fig. 5A, B). The main outcomes in terms of cartilage formation (p < 0.01) and detection of PDX1+ cells (p < 0.001) and of PDX1-NKX6.1 double-positive cells (p < 0.05) are summarized in Table 3.
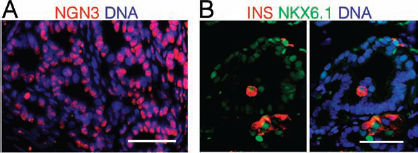
Immunofluorescence analysis of subcutaneous grafts at 12 weeks posttransplantation. (A) At 12 weeks posttransplantation, a group of NGN3+ cells were observed in the subcutaneous graft. (B) Scattered insulin-positive (INS) cells that coexpressed NKX6.1 could be detected in the subcutaneous implants. Scale bar: 50 μm.
Distribution of Graft Outcomes at Early and Late Time Points.
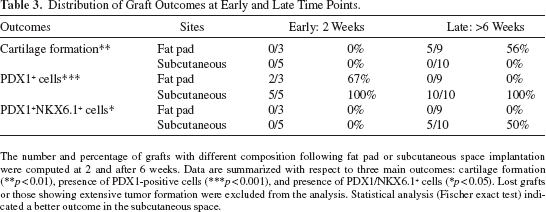
The number and percentage of grafts with different composition following fat pad or subcutaneous space implantation were computed at 2 and after 6 weeks. Data are summarized with respect to three main outcomes: cartilage formation (**p < 0.01), presence of PDX1-positive cells (***p < 0.001), and presence of PDX1/NKX6.1+ cells (*p < 0.05). Lost grafts or those showing extensive tumor formation were excluded from the analysis. Statistical analysis (Fischer exact test) indicated a better outcome in the subcutaneous space.
In all grafts, at all time points, we noticed frequent blood vessels containing red blood cells. There were no obvious differences with respect to the density of these vessels in the grafts. These results obtained between 2 and 12 weeks posttransplantation suggest that PDX1+ cells derived from hESCs by our culture method survived and further developed along the pancreatic lineage much better in the subcutaneous than in the epididymal fat pad site. Only in the subcutaneous site, these PPP cells further differentiated into PDX1-, SOX9-, NKX6.1-, and NGN3-expressing cells that can be considered as fully specified pancreatic progenitors and endocrine progenitors.
Discussion
Recent studies on the derivation of pancreatic cells from human pluripotent stem cells have generated much excitement in the field and hope of new treatment options for type 1 diabetes. A first breakthrough was the in vitro generation of pancreatic endoderm (5,10) and, later, of functional endocrine cells derived thereof following ectopic transplantation in mice (12). However, several reports appeared thereafter that the in vitro protocol could not be reproduced in other laboratories with different human embryonic stem cells (17,22). Similarly, the in vivo “maturation” into functional endocrine cells could not be reproduced when pancreatic endoderm obtained from the original protocol was transplanted in the fat pad of rats (15).
We recently reported a differentiation protocol that reproducibly derives pancreatic endoderm from various hESC lines (17). In the present study, we examined the in vivo fate (survival, proliferation, and differentiation) of these cells following transplantation in the fat pad or subcutaneously. Unexpectedly, we found a site-specific differentiation of the transplanted hESC-derived pancreatic endoderm cells. At the subcutaneous site, the transplanted cells continued to stably express PDX1 and other pancreatic endoderm markers for the entire 12-week duration of the experiment. These cells showed epithelial morphology and were arranged in cystic or duct-like structures that can be considered as reminiscent of early pancreatic morphogenesis. Other major transcription factors regulating pancreatic endocrine differentiation became expressed, namely NKX6.1 from 6 weeks and NGN3 from 12 weeks. This indicates further specification along the pancreatic lineage at the subcutaneous site. However, in the fat pad site, only few PDX1+ cells remained after 2 weeks, and they were completely lost from 6 weeks. At this site, the epithelium was replaced by mesenchyme and even cartilage that had probably formed from the contaminating nonendodermal cells. Cartilage formation was never observed in the subcutaneous site.
Our observations indicate that the subcutaneous site is a more permissive environment for pancreatic lineage survival, proliferation, and differentiation whereas the fat pad promotes chondrocytic (mesodermal) development. It is currently not known why the transplanted hESC-derived cells responded differently at these two transplantation sites. Probably, the fat pad does not provide the necessary survival and proliferation factors that, however, are present in the subcutaneous site. What could these factors be? Histological examination of the grafts revealed no obvious differences with respect to vascularization. So, oxygen, nutrients, or other factors supplied by blood vessels could not explain the observed differences between subcutaneous and fat pad graft composition. Adipose tissue seems to produce a different environment, with different paracrine signals, than the subcutaneous connective tissue. For example, insulin-like growth factor-1 (IGF-1) and transforming growth factor-β (TGF-β) are factors produced by adipocytes that are known to promote chondrogenesis (4,26). Hedgehog (Hh) signaling emerges as a potential mediator of fat formation during postnatal development and is expressed in adipose as well as epididymal tissue (7,25). However, Hh is strongly inhibitory to pancreatic differentiation (16,18). The subcutaneous space has also been shown to be permissive for pancreatic differentiation from fetal pancreatic explants (8). In the scope of this preferential survival and growth in the subcutaneous site, one should also consider the recent demonstration of similarities (phenotype and differentiation potentials) between skin stem cells and pancreatic stem cells (11). Whether these skin stem cells contribute to our observations is not known. But if yes, it should be indirectly since PDX1+ cells in grafts were shown to be of human origin (Hu Mitochondria positive).
In the subcutaneous environment and in a 6 weeks time, our hESCs-derived PPP cells proliferated and became multimarkers positive cells (PDX1, FOXA2, SOX9, CK19, and NKX6.1) that we refer to as fully specified pancreatic progenitors. Furthermore, they generated NGN3+ endocrine progenitors as well as some insulin+ cells between 6 and 12 weeks posttransplantation. As only a marginal number of insulin+ cells was detected, we did not perform any functional test to characterize their degree of maturation. The low proportion of endocrine cells detected here is comparable to previous work from other groups. Recently, Matveyenko et al. (15) transplanted hESC-derived PDX1+/NGN3+ cells in nude rats but detected only a small area of insulin-positive cells in 50% of the grafts after a period of 20 weeks. Additionally, Eshpeter et al. (6) and Phillips et al. (21) could not detect any insulin-expressing cells until 6 weeks posttransplantation, and the transplanted diabetic animals did not show any significant reduction in blood glucose levels. We assume that it would be necessary to prolong the transplantation time in order to allow further differentiation of the NGN3+ progenitors into mature endocrine cells.
It remains a major challenge how these pancreatic progenitors can be harnessed to become mature, functional β-cell equivalents. There is currently a knowledge gap concerning the extracellular factors driving terminal differentiation of β-cells during development. The subcutaneous site is an easily accessible site that would allow administration of putative β-cell-inducing factors to be tested. This model opens new prospects to the identification of such factors. It also offers a technical advantage over other sites. For instance, the total surface area available for transplantation is large; the grafted cells are easily accessible and can be retrieved at several intervals from the same animal, therefore eliminating the need for invasive surgery and its related risks.
Footnotes
Acknowledgments
This work was supported by a Vrije Universiteit Brussel (VUB) research grant under the code GOA41. The authors declare no conflicts of interest.