
Abstract
We previously reported the in vitro differentiation of human embryonic stem cells (hESCs) into pancreatic endoderm. Here we demonstrate that islet-like three-dimensional (3D) aggregates can be derived from the pancreatic endoderm by optimizing our previous protocol. Sequential treatment with Wnt3a, activin A, and noggin induced a transient upregulation of T and MixL1, followed by increased expression of endodermal genes, including FOXA2, SOX17, and CXCR4. Subsequent treatment with retinoic acid highly upregulated PDX1 expression. We also show that inhibition of sonic hedgehog signaling by bFGF/activin βB and cotreatment with VEGF and FGF7 produced many 3D cellular clusters that express both SOX17 and PDX1. We found for the first time that proteoglycans and vimentin+ mesenchymal cells were mainly localized in hESC-derived PDX1+ clusters. Importantly, treatment with chlorate, an inhibitor of proteoglycan sulfation, together with inhibition of Notch signaling significantly increased the expression of Neurog3 and NeuroD1, promoting a transition from PDX1+ progenitor cells toward mature pancreatic endocrine cells. Purified dithizone+ 3D aggregates generated by our refined protocol produced pancreatic hormones and released insulin in response to both glucose and pharmacological drugs in vitro. Furthermore, the islet-like 3D aggregates decreased blood glucose levels and continued to exhibit pancreatic features after transplantation into diabetic mice. Generation of islet-like 3D cell aggregates from human pluripotent stem cells may overcome the shortage of cadaveric donor islets for future cases of clinical islet transplantation.
Introduction
Type 1 diabetes mellitus (DM) results from the autoimmune destruction of insulin-producing β-cells in pancreatic islets, and exogenous insulin injection has been the major therapeutic option to control blood glucose in such patients (1). However, patients with type 1 DM still suffer from diabetic complications due to the lack of physiological oscillations in insulin secretion. Thus, the most effective and physiological way to treat DM is to restore the functional β-cell mass so that it can synthesize and secrete insulin in response to changing glucose concentrations in the blood. Transplantation of either the whole pancreas or isolated islets has shown positive outcomes, resulting in considerable improvements in morbidity (1). However, despite an increasingly urgent need, the scarcity of cadaveric donor islets and difficulties in expanding differentiated β-cells in vitro are still problematic.
Human pluripotent stem cells, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), have been proposed as promising renewable sources of many different cell types for cell replacement therapy, disease modeling, and drug screening (36,50). Our previous studies showed that brain, pancreas, and hepatic functions can be restored by transplantation of differentiated mouse or human ESCs (hESCs) and iPSCs (21,22,43,49). During the past decade, considerable progress has been made to generate insulin-producing β-cells from ESCs and iPSCs using developmentally relevant recombinant proteins and small molecules (3,13,33,48,51). However, in most cases, β-cells derived from hESCs in vitro exhibit immature pancreatic endocrine functions, showing a limited response to glucose stimulation (4,33,48). In addition, in vivo studies showed that robust glucose-induced insulin secretion could be obtained in hESC-derived β-cells only after several months of grafting (4,18,41,43). Thus, there is a clear need for more robust and improved differentiation procedures before hESC-based approaches can become therapeutic options for treating diabetes.
Heparan sulfate proteoglycans (HSPGs) are localized at the cell surface and in extracellular matrix and modulate the action of receptor-signaling modules by interacting with a variety of ligands. Previous studies provided direct evidence that pancreatic mesenchyme in developing rat and mouse embryos promoted progenitor cell proliferation but inhibited differentiation into endocrine cells (5,30). A subsequent study showed that sulfate proteoglycans are produced in the mesenchyme surrounding the pancreatic epithelium and negatively regulate endocrine differentiation in rat embryonic pan-creata (52). Therefore, these data suggest potential negative roles of proteoglycans in β-cell differentiation of human pluripotent stem cells. However, previous studies mainly focused on the treatment with exogenous inductive signals, but few attempts have been made to eliminate inhibitory signals that may be produced from different types of cells differentiating from human pluripotent stem cells.
The topological arrangement or intercellular communication of differentiating endocrine cells may be important for regulated insulin secretion (11,13,35), but has been often overlooked in previous studies. Therefore, the development of methods based on the understanding of the three-dimensional (3D) cellular architecture of pancreatic islets may be an ideal approach to generate more functional β-cells in a large number.
We previously demonstrated that the differentiation of pancreatic progenitors can be efficiently directed from hESCs by sequential exposure to epigenetic signals (43). In this study, we developed a refined protocol for differentiating hESCs into functional islet-like 3D cellular spheroids that produce pancreatic hormones and respond to changing glucose levels both in vitro and in vivo. Furthermore, we show for the first time that inhibition of proteoglycan sulfation significantly increases Neurog3 and NeuroD1 expression, promoting endocrine differentiation of hESC-derived PDX1+ pancreatic progenitor cells.
Materials and Methods
Culture and Differentiation of hESCs
hESC lines (HSF6, female, NIH code UCO6, passages 55–65; H1, male, NIH code WA01, passages 25–35; Miz-hES6, female, Korean Stem Cell Research Center registered cell line, passages 60–90) were maintained on a feeder layer of mitomycin C (Roche, Mannheim, Germany)-treated mouse embryonic fibroblasts isolated from 13.5-day-old CF-1 mice embryos (Orient Bio, Seoul, Korea) in a mixture of Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL, Grand Island, NY, USA) and Ham's F-12 medium (1:1; Gibco-BRL) supplemented with 20% KnockOut serum replacement (Gibco-BRL), 1 mM nonessential amino acids (Invitrogen, Grand Island, NY, USA), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA), 100 U/ml penicillin G (Gibco-BRL), 100 μg/ml streptomycin (Gibco-BRL), and 4 ng/ml human basic fibroblast growth factor (bFGF; R&D Systems, Minneapolis, MN, USA). Every 5–6 days, hESC colonies were subcultured by treatment with 1 mg/ml collagenase type IV (Invitrogen). hESCs were incubated in a standard gas atmosphere containing humidified 5% CO2 and air (21% O2) at 37°C. All experimental procedures involving hESCs were approved by the institutional review board of the Korea University (1040548-KU-IRB-13-9-A-1).
Stage I: Definitive Endoderm/3D Aggregate Formation
Pancreatic differentiation was initiated by treating hESCs sequentially with Wnt3a (R&D Systems), activin A (R&D Systems), noggin (R&D Systems), and all-trans RA (Sigma-Aldrich) during human embryoid body (hEB) formation. During the first 2 days, hEBs were cultured in the presence of Wnt3a and activin A (both 30 ng/ml). Subsequently, hEBs were treated with activin A and noggin (both 30 ng/ ml) for the next 4 days, and with RA (10 μM) and 30 ng/ml noggin for the final 2 days.
Stage II: Pancreatic Endoderm/2D Adherent Culture
Eight-day-old hEBs were dissociated and cultured in DMEM containing B27 (Gibco-BRL) and 10 mM nicotin-amide (NA; Sigma-Aldrich) on Matrigel (R&D Systems)-coated tissue culture dishes for 6 days. During this stage, dissociated hEBs were treated with 1 ng/ml bFGF, 50 ng/ml activin βB (R&D Systems), 30 ng/ml vascular endothelial growth factor (VEGF; R&D Systems), 100 μM ascorbic acid (Sigma-Aldrich), and 100 μM trolox (Sigma-Aldrich). Additionally, 30 ng/ml FGF7 (R&D Systems) was administered for the last 2 days.
Stage III: Pancreatic Endocrine Precursor Cells/2D Adherent Culture
Cells were cultured on Matrigel-coated dishes in CMRL-1066 (Gibco-BRL) containing B27 supplement (Gibco-BRL), 10 mM NA, and 100 mM sodium chlorate (Sigma-Aldrich) for 4 days. Additionally, 1 μM g-secretase inhibitor (GSI; Calbiochem, Darmstadt, Germany), and 1 ng/ml transforming growth factor-β1 (TGF-β1; R&D Systems) were added. The concentration of glucose (Sigma-Aldrich) in the culture medium was 17.5 mM during stages III and IV.
Stage IV: Hormone-Expressing Endocrine Cells/3D Aggregate Culture
At the onset of stage IV, differentiated pancreatic endocrine cells were enriched by handpicking dithizone (DTZ; Sigma-Aldrich)-positive clusters without trypsin under a stereomicroscope (Carl Zeiss, Jena, Germany) and further differentiated for 6 days in suspension culture. The basal medium used during stage IV was CMRL-1066 medium with B27 supplement plus 50 ng/ml exendin-4 (Sigma-Aldrich), 10 mM NA, 30 ng/ml hepatocyte growth factor (HGF; ProSpec, East Brunswick, NJ, USA), and 30 ng/ml insulinlike growth factor 1 (IGF-1; R&D Systems).
Fluorescence-Activated Cell Sorting Analysis
Cells in stage I were fixed and permeabilized with the Cytofix/cytoperm solution (Becton Dickinson, San Diego, CA, USA). Cells were then labeled with anti-human SOX17 antibody (1:50, 562205; BD Biosciences, San Jose, CA, USA), followed by fluorescence-conjugated second ary antibodies (1:200, A21202; Invitrogen). Fluorescence-activated cell sorting Calibur (FACS Calibur) with CellQuest software (Becton Dickinson) was used to quantify the immunoreactive cells.
RNA Extraction and RT-PCR Analysis
Total RNA was isolated using the TRIzol reagent (Invitrogen), and cDNA was synthesized from 2 μg total RNA using the SuperScript III First-Strand Synthesis kit (Invitrogen). PCR was subsequently carried out using AccuPower PCR-Premix (Bioneer, Daejeon, Korea). The primer sequences and reaction conditions are listed in Table 1. Relative band intensities were determined using an AutoChemi system (UVP BioImaging Systems, Upland, CA, USA). The levels of target mRNAs were normalized to the signal obtained for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression (Table 1). FirstChoice Human Pancreatic Total RNA (Ambion, Austin, TX, USA) was used as a positive control.
Primers and Reaction Conditions for RT-PCR

Real-Time Quantitative PCR
Real-time quantitative RT-PCR was performed using iQTM SYBR® Green Supermix (Bio-Rad, Hercules, CA, USA) expression assays on differentiated cells according to the manufacturer's instructions (primer sequences are listed in Table 2; Applied COSMO Genetech, Seoul, Korea). Briefly, 20 μl of the RT-PCR mixture contained 10 μl 2× iQTM SYBR® Green Supermix, 50 ng cDNA, and 1 μl of 20× SYBR® Green expression assays. The CT of the target genes was analyzed using the CFX-96 real-time PCR system (BIO-RAD). The comparative CT method (2–δδCt) was used to determine the relative quantification of the target genes normalized to GAPDH (Table 2).
Primers and Reaction Conditions for qPCR

Western Blotting Analysis
Cells were harvested in RIPA lysis buffer (Upstate, Charlottesville, VA, USA) supplemented with a cocktail tablet of protease inhibitors (Roche). Equal amounts of protein (50 μg/lane) were electroporated on NuPageTM 4–12% Bis-Tris polyacrylamide gels (Invitrogen) and transferred onto PVDF membranes (Invitrogen). Membranes were incubated with primary antibodies against SOX17 (1:400, AF1924; R&D Systems), PDX1 (1:200, AF2419; R&D Systems), Neurogenin 3 (1:100, sc-23832; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), NeuroD1 (1:500, sc-1084; Santa Cruz Biotechnology), and β-actin (1:200, sc-1615; Santa Cruz Biotechnology) overnight at 4°C. The membranes were then rinsed with TBS-T and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (1:10,000, sc-2020; Santa Cruz Biotechnology) for 1.5 h at room temperature. Immunoreactive proteins were detected using ECL Plus reagents (GE Healthcare, Pittsburg, PA, USA).
Immunofluorescence Staining
Cells and tissues were fixed in cold 4% paraformaldehyde (PFA; Sigma-Aldrich) in phosphate-buffered saline (PBS, Gibco-BRL). Tissues were embedded in OCT compound (Tissue Tek®, Sakura, Japan) after fixation and were sectioned into 8- to 10-μm slices. After blocking and permeabilization in 0.3% Triton X-100 (Sigma-Aldrich) and 10% donkey serum (Sigma-Aldrich) in 0.1% bovine serum albumin (BSA; Sigma-Aldrich)/PBS, the cells or tissue sections were incubated at 4°C overnight with primary antibodies against SOX17 (1:100; R&D systems), PDX1 (1:100; R&D systems), FOXA2 (1:1,000, sc-6554; Santa Cruz Biotechnology, and 1:10, 4C7; Developmental Studies Hybridoma Bank, Iowa City, IA, USA), HNF1α (1:500, sc-6548; Santa Cruz Biotechnology), e-cadherin (sc-7870, 1:200; Santa Cruz Biotechnology), keratin 19 (1:100, M0888; Dako, Glostrup, Denmark), vimentin (1:500, M 0725; Dako), somatostatin (1:300, A0566; Dako), insulin (1:500, 4011-01F; Linco, St. Charles, MO, USA), c-peptide (1:200, 4020-01; Linco), proinsulin (1:100, 10-P14; Fitzgerald, Concord, MA, USA), glucagon (1:400, G2645, Sigma-Aldrich), and human nuclei (1:200, MAB1281; Chemicon, Temecula, CA, USA). Samples were then washed with PBS and probed with appropriate fluorescence-tagged secondary antibody (1:400, A21207, A21230, A11016, A21202, A11055; Invitrogen) in 0.1% BSA/PBS at room temperature. Normal human pancreas sections (Abcam, Cambridge, MA, USA) were used as positive controls for immunostaining of pancreatic hormones. For in vitro proliferation assay, cells were incubated with 5-ethynyl-2′-deoxyuridine (EdU) for 1 h and processed using a Click-iT® EdU imaging Kit (Invitrogen) according to the manufacturer's instructions. Apotome-Axiovert 200 M fluorescence microscope (Carl Zeiss) and confocal laser scanning microscope (Carl Zeiss) were used to visualize cells after counterstaining with 4′,6-diamidino-2-phenylindole (DAPI; 1:100, 100-43-6; Sigma-Aldrich).
Dithizone and Alcian Blue Staining
A DTZ stock solution was prepared as a solution of 10 mg/ml DTZ in dimethyl sulfoxide (Sigma-Aldrich) and stored at -15°C in the dark. The staining solution was freshly prepared by the addition of 10 μl stock solution into 1 ml PBS and filtration through a 0.2-μm-pore syringe filter (Sartorius Inc., Goettingen, Germany). hESC-derived pancreatic endocrine cells were incubated with the DTZ staining solution at 37°C for 15 min and washed with PBS. Positive staining was observed as crimson-red clusters with an Axiovert stereomicroscope (Zeiss). For Alcian blue staining, cells in stage III were fixed with 4% PFA for 30 min, rinsed with PBS, and stained with 1% Alcian blue solution (Gibco-BRL) prepared in 0.1 N HCl (Sigma-Aldrich).
Insulin Content and Secretion Assays
DTZ+ cell clusters were harvested, washed four times, and incubated in 3.3 mM glucose containing RPMI-1640 supplemented with 10 mM HEPES buffer (Gibco-BRL) and 10% fetal bovine serum (Gibco-BRL) for 1 h. Pancreatic endocrine cells differentiated from hESCs were incubated for 2 h at 37°C in the presence of either 3.3 mM or 25 mM glucose (Sigma-Aldrich). Different agonists and antagonists of signaling pathways for insulin secretion were added, including diazoxide (500 μM), carbachol (10 μM), tolbutamide (10 μM), 3-isobutyl-1methylxanthine (IBMX, 100 μM), and nifedipine (50 μM) (Sigma-Aldrich). The supernatant was harvested and analyzed by insulin ELISA kit (Dako) according to the manufacturer's instructions. To normalize the amount of insulin secretion, the total protein concentration of the cells in each condition was measured by the Bradford method.
Induction of Diabetes and Transplantation of hESC-Derived Pancreatic Endocrine Cells
Eight- to 10-week-old male nude mice (Charles River, Wilmington, MA, USA) were used as recipients for implantation (control, n = 4; sham, n = 4; hESC-derived pancreatic endocrine cells, n = 6). DM was induced by intraperitoneal injection of streptozotocin (STZ; Sigma-Adrich) at a concentration of 200 mg/kg body weight. Before implantation, DM status was checked every other day by body weight and blood glucose levels. After 4–6 days of STZ treatment, differentiated pancreatic endocrine cells (3 × 106) cells were transplanted into diabetic mice (blood glucose ≥ 400 mg/dl) by injection into the kidney capsule. The control animals did not receive any implantation, and sham mice received only an injection of tissue culture medium used to support the cell suspension. Blood glucose levels and body weights were monitored under nonfasting conditions for 110 days. Blood glucose levels were measured with a portable glucometer (Accu-chek Active; Roche, Basel, Switzerland). The kidneys were dissected and fixed in 4% PFA for immunostaining. For the intraperitoneal glucose tolerance test (IPGTT), mice were fasted for 16 h and given an intraperitoneal injection of glucose (2 g/kg body weight). Venous blood was obtained from the tail vein 0, 30, 60, 90, 120, 150, and 180 min after the glucose injection. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Korea University (KUIACUC-2010-143).
Statistical Analysis
All statistical analysis was performed using SAS software (version 8.2; SAS Institute, Inc., Cary, NC, USA). Data shown are mean values of at least three independent experiments performed in triplicate. Group means were analyzed using Student's t-test for pairwise comparisons. For multiple comparisons, one-way ANOVA followed by Tukey's test was used. Significant differences between several groups were reported when p < 0.05.
Results
Differentiation Into Endodermal Cells
We previously reported that PDX1+ pancreatic endoderm can be generated from hESCs by sequentially exposing hEBs to serum, activin A, and RA (43). In the present study, we further optimized and improved this protocol by testing different combinations of growth factors and culture media; a schematic of the differentiation conditions is shown in Figure 1a. During the first 2 days of stage I (hEB formation), we substituted serum with Wnt3a (30 ng/ml) (Fig. 1a), which is known to promote the formation of mesendoderm while suppressing the neuroectodermal fate (31,45). In addition, noggin (30 ng/ ml), which induces the formation of posterior foregut endoderm (38), was administered in serum-free conditions for an additional 6 days (Fig. 1a). hEBs were treated with activin A (30 ng/ml) during the first 4 days, and RA (10 μM) was added for the last 2 days of EB formation as per our previous protocol (43). Real-time quantitative PCR (qPCR) was carried out to analyze the gene expression associated with hESCs, mesendoderm, and definitive and pancreatic endoderm during the 8 days of stage I (Fig. 1b). The results showed a time-dependent decrease in the gene expression levels of the hESC marker, POU domain class 5 transcription factor 1 [POU5F1, also known as octamer-binding transcription factor-4 (OCT4)], over the 8 days of hEB formation. The gene expression levels of mesendodermal markers T (Brachyury) and Mix 1 homeobox-like 1 (MIXL1) were immediately and transiently increased 1 day after combined treatment with Wnt3a and activin A, but decreased over the next 5 days (Fig. 1b). The reduction of T and MIXL1 expression levels occurred concomitantly with increases in the expression levels of endodermal genes, FOXA2, SOX17, and C-X-C chemokine receptor type 4 (CXCR4). Upon the addition of RA, the cells rapidly began to express high levels of PDX1 (Fig. 1b). Flow cytometric analysis of cells at day 6 of differentiation showed that the number of SOX17+ cells was highly increased by the treatment with Wnt3a, activin A, and noggin, compared to control differentiation without growth factors (Fig. 1c).

Schematic and analyses of pancreatic differentiation. (a) Schematic representation of the culture conditions in each stage of the differentiation procedure. Note that cells were grown as 3D spheroids in stages I and IV. (b) qPCR analysis of gene expression during stage I of hEB formation. RT-PCR was performed using RNA from undifferentiated hESCs or cell aggregates collected at the indicated time points to quantify POU5F1, NANOG, MIXL1, FOXA2, SOX17, PDX1, and CDX2 gene expression. The data represent the means ± SD of four separate experiments. (c) Flow cytometric analysis of SOX17-expressing cells (47.8%) at day 6 of stage I (red). Blue, isotype control; Green, control differentiation without growth factors (7.5%). Data shown are representative histograms from three independent experiments.
Differentiation Into Early Pancreatic Progenitor Cells
Interactions of the developing gut tube with the neighboring notochord, dorsal aorta, and lateral plate mesoderm provide pancreatic inductive signals and initiate pancreatic budding (27,29). For further differentiation into pancreatic endoderm, 8-day-old hEBs (stage I) were dissociated and cultured in DMEM/B27 containing notochord and vascular signals (i.e., bFGF, activin bB, and VEGF) on Matrigel-coated culture dishes for an additional 6 days (stage II, Fig. 1a). Nicotine amide (NA, 10 mM), which is a potential inducer of β-cell outgrowth in the human fetal pancreas (39), was also included in the culture medium. Under this culture condition, many cells coexpressed the primitive gut tube and ductal epithelial markers E-cadherin, SOX17, FOXA2, keratin 19 (K19), and hepatocyte nuclear factor-1 homeobox A (HNF1A) (Fig. 2a–c). Interestingly, many cell aggregates were readily observed in stage II (Fig. 2d), and these aggregates that were budded from the adherent cell monolayer expressed SOX17 and PDX1 (Fig. 2e, f).
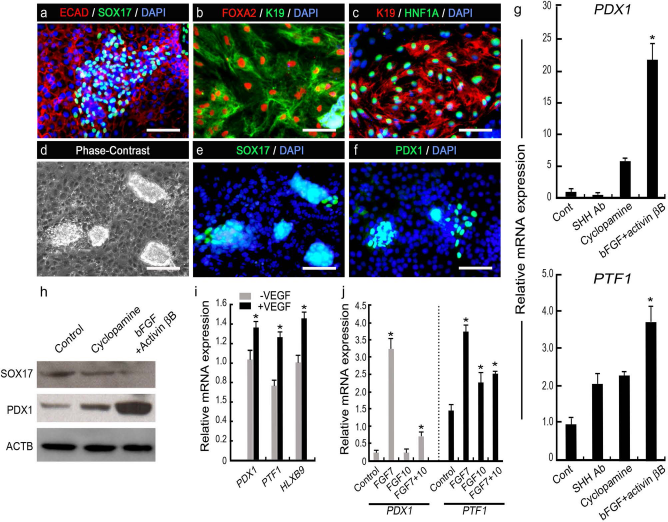
Differentiation into pancreatic endoderm from hESCs. (a–c) Representative immunofluorescence images of dissociated hEBs, showing the expression of markers associated with primitive gut tube or pancreatic endoderm (ECAD, SOX17, FOXA2, K19, and HNF1A). (d–f) Phase contrast and immunofluorescence images of cell clusters produced in stage II. Note that the clusters expressed SOX17 and PDX1. (g) Relative gene expression PDX1 and PTF1A following treatments with SHH-neutralizing antibody (SHH Ab), SHH signaling inhibitor cyclopamine, or bFGF + activin βB in DMEM containing B27 and NA. The expression levels of the target genes were normalized to GAPDH. Data represent the means ± SD of triplicate experiments. *p < 0.05 vs. control cells. (h) Western blot analysis of stage II cells for SOX17 and PDX1 after treatment with cyclopamine or bFGF + activin βB. ACTB, β-actin. (i) The expression levels of PDX1, PTF1A, and HLXB9 in stage II cells grown in the absence (-) and presence (+) of VEGF. *p < 0.05 versus -VEGF. (j) Relative expression levels of PDX1 and PTF1 after treatment with FGF7, FGF10, or both FGF7 and FGF10. *p < 0.05 versus control cells grown in DMEM containing B27, NA, bFGF, and activin βB. Scale bars: 100 μm.
Inhibition of SHH expression in the developing gut tube endoderm permits the expression of normal pancreas genes, including PDX1 and INS (14). To repress SHH signaling in stage II, we first tested notochord signals such as bFGF and activin bB, a hedgehog signaling inhibitor cyclopamine, and SHH-neutralizing antibody in DMEM/ B27 containing 10 mM of NA, and comparatively analyzed the induction of PDX1 and pancreas transcription factor 1 subunit a (PTF1A) gene expression levels at the end of stage II. The combined treatment of differentiating hESCs with bFGF and activin βB during the 6 days of stage II most effectively increased the expression levels of PDX1 and PTF1A compared to treatment with SHH antibody and cyclopamine (Fig. 2g). Notably, Western blot analysis showed that the expression of SOX17 protein was lower after bFGF/activin βB addition than after cyclopamine and control treatment, but PDX1 expression was profoundly induced by bFGF/activin βB treatment at the protein level (Fig. 2h).
We next tested FGF7, FGF10, and VEGF (all 30 ng/ ml) in the presence of bFGF and activin βB during the 6 days of stage II to mimic the in vivo endothelial or mesenchymal signaling during early pancreas development (28). Studies of the different durations of administration showed that 6 days of VEGF treatment and the last 2 days of FGF7 treatment most effectively induced the expression of PDX1, PTF1, and/or HLXB9 (Fig. 2i, j).
Differentiation Into Pancreatic Endocrine Cells
Epithelial–mesenchymal interaction is critical for the differentiation of pancreatic endocrine cells in the developing pancreas. Previous studies demonstrated that mesenchymal cells support the proliferation of progenitor cells but suppress endocrine differentiation by delaying the expression of neurogenin 3 (neurog3), which is a key transcription factor in the specification of pancreatic endocrine cells (5,8). The inhibitory effect was mediated by direct epithelial–mesenchymal contact and required the Notch signaling pathway (5). Based on these findings, we first localized the mesenchymal cells using antibody against vimentin, which is known to be expressed in pancreatic mesenchyme (20,30). At the end of stage II, a number of PDX1+ clusters were observed, and many cells surrounding and inside clusters were labeled with EdU (Fig. 3a). In stage III, differentiating hESCs were grown in CMRL-1066 medium, and the concentration of glucose in the culture medium was reduced from 25 mM to 17.5 mM to avoid the potential high glucose-induced impairment of the functional maturation of β-cells in vitro (44). Interestingly, vimentin+ mesenchymal cells are preferentially distributed within PDX1+ clusters (Fig. 3b), suggesting potential influences of mesenchymal cells on proliferation and differentiation of PDX1+ cells. Confocal imaging of the clusters revealed that the vimentin expression was mainly detected in PDX1– cells surrounding the cluster (Fig. 3c, upper left and bottom right panels, c3). Interestingly, however, vimentin was also detected in some of cells expressing low level of PDX1 at the periphery of clusters (Fig. 3c, c1), while cells showing strong signals of PDX1 in the center did not express or express vimentin at very low level (Fig. 3c, c2). A recent study showed that sulfated proteoglycans located in mesenchyme negatively regulate endocrine differentiation (52). RT-PCR analyses revealed that two main proteoglycan families, syndecans and glypicans, and enzymes involved in N-decacetylation and sulfation of glycosaminoglycan chains (NDST1, HS2ST1, HS6ST1, and HS6ST2) were all expressed in differentiating hESCs (Fig. 3d). In addition, Alcian blue staining showed that high-sulfate proteoglycans were accumulated mainly in the clusters (Fig. 3e). Thus, we hypothesized that treating differentiating hESCs in stage III with chlorate, an inhibitor of proteoglycan sulfation, might increase the expression of Neurog3, promoting endocrine differentiation. Treatment of differentiating hESCs in stage III with sodium chlorate significantly increased Neurog3 expression by more than twofold (Fig. 3f). In addition, the chlorate treatment upregulated the expression of NeuroD1, an insulin gene transactivator, which is known to be activated by neurog3 and required to achieve and maintain functional maturity of β-cells (10) (Fig. 3f). Western blot analyses showed that sodium chlorate increased the level of both Neurog3 and NeuroD1, while it decreased PDX1 expression (Fig. 3g).
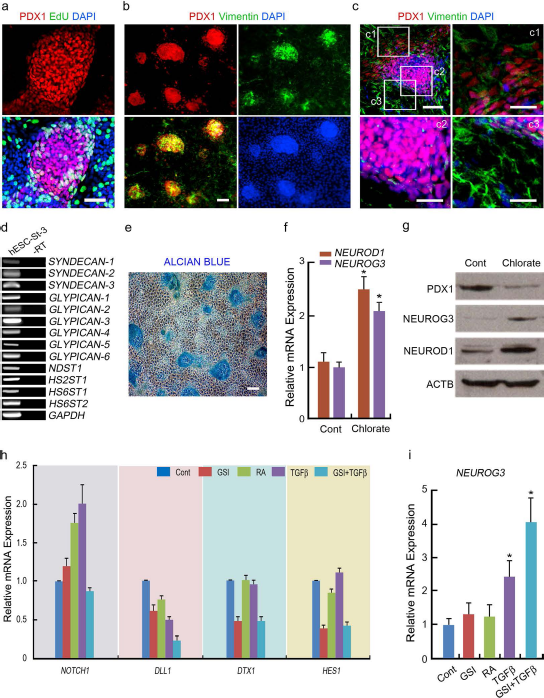
Enhancement of endocrine differentiation by inhibiting Notch and mesenchymal signals. (a) Immunofluorescence images of a cell cluster for PDX1 and EdU at the end of stage II (day 14 of differentiation). Scale bars: 50 μ. (b) Immunofluorescence images of clusters for PDX1 and vimentin in stage III. Scale bars: 100 μ. (c) Confocal images of immunofluorescent staining for PDX1 and vimentin in cell clusters. Scale bars: 100 μ. Boxed areas (c1, c2, and c3) in the upper left are shown as enlarged images in the upper right and bottom (Scale bars: 50 mm). (d) RT-PCR analysis of stage III cells for syndecan-1–3; glypican-1–6; and N-deacetylase/ N-sulfotransferase-1 (NDST1), heparin sulfate-2-sulfotransferase (HS2ST1), and heparin sulfate-6-sulfotransferase 1 and 2 (HS6ST1 and HS6ST2). (e) Detection of sulfate proteoglycans in cells of stage III by Alcian blue staining. Scale bars: 100 μ. (f) Expression of NEUROG3 and NEROD1 in stage III cells differentiated in the presence (Chlorate) or absence (Cont) of sodium chlorate (100 mM). (g) Western blot analyses for PDX1, Neurog3, and NeuroD1 in stage IIl cells treated with (Chlorate) or without (Cont) sodium chlorate. (h, i) Real-time quantitative PCR analysis of gene expression levels associated with Notch signaling (NOTCH1, DLL1, DTX1, and HES1) and pancreatic endocrine differentiation (Neurog3) after treatment with GSI, RA, TGF-β1, or both GSI and TGF-β1. Control cells were grown in CMRL-1066 medium containing B27, NA, and sodium chlorate. Data represent the means ± SD of three independent experiments performed in triplicate. *p < 0.05 versus control cells.
Neurog3 expression is induced in endocrine precursors through the inhibition of the Notch downstream transcription factor hairy and enhancer of split-1 (HES1) (17). Thus, in the presence of sodium chlorate, we evaluated the expression of Notch-related genes upon treatment with Notch inhibitor GSI (1 μM), TGF-β1 (1 ng/ml), and RA (10 μM), which are known to induce the differentiation and proliferation of endocrine cells (42,46,47) (Fig. 3h). Gene expression analyses showed that RA and TGF-β1 had either no or a minimal effect on the downregulation of notch homolog 1 (NOTCH1), delta-like 1 (DLL1), del-tex1 (DTX1), and HES1 (Fig. 3h). In contrast, GSI alone, and in combination with TGF-β1, significantly decreased the expression of Notch-associated genes (Fig. 3h). In line with this observation, combined treatment with GSI and TGF-β1 in stage III dramatically increased Neurog3 expression (Fig. 3i).
Maturation of Dithizone+ Islet-Like 3D Clusters
At the end of stage III, a large area of the culture dishes was covered by clusters of cells that had budded from the adherent monolayer. These clusters were positive for DTZ, a zinc chelator known to selectively stain β-cells in the pancreatic islets (Fig. 4a) (44). The number of DTZ+ clusters was profoundly increased by our protocol compared to spontaneously differentiated control cultures (Fig. 4a). In stage IV, DTZ+ clusters were collected by mechanical detachment after gentle pipetting or handpicking without trypsin and allowed to mature for 6 days in suspension culture (Fig. 4b). To promote the survival and differentiation of pancreatic endocrine cells and prevent apoptosis, the hESC-derived DTZ+ clusters were cultured in CMRL-1066 medium containing IGF-1 (30 ng/ml), HGF (30 ng/ml), and exendin-4 (50 ng/ml) (4,25,26). After 6 days of maturation, islet-like spheroids coproduced INS, proinsulin (pro-INS), and c-peptide (c-pep), as determined by immunofluorescence staining (Fig. 4c, left and middle panels; human pancreatic tissues were used as positive controls, right panels). The percentage of INS+ cells varied considerably between the clusters, and some clusters contained only a few INS+ cells (Fig. 4d, e, upper and middle panels; human pancreatic tissues were used as positive controls, bottom panels). In addition, a small subset of cells in the spheroids produced glucagon (GCG) and somatostatin (SST).
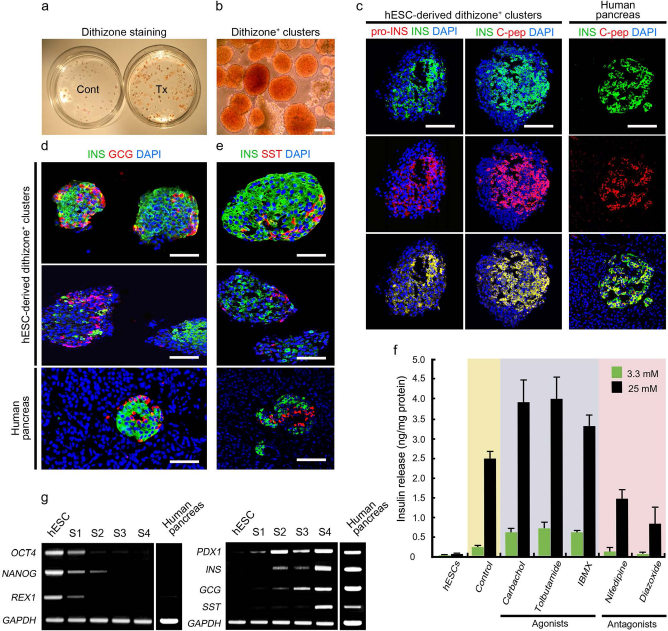
Enrichment and characterization of hESC-derived islet-like aggregates. (a) DTZ staining of differentiated hESCs. At the end of stage III (day 18 of differentiation), the number of DTZ+ clusters was markedly increased (Tx) compared to the control spontaneous differentiation for the same duration (Cont). (b) DTZ+ aggregates after isolation by mechanical detachment. (c–e) Immunofluorescence labeling of DTZ+ aggregates and human pancreatic tissue sections with anti-INS, -pro-INS, -c-pep, -GCG, and -SST antibodies. All cells in aggregate and tissue sections were counterstained with DAPI. (f) Levels of insulin secretion in low (3.3 mM) and high (25 mM) glucose conditions and after treatment with multiple insulin agonists and antagonists. Control, DTZ+ clusters in the absence of agonists and antagonists. (g) The expression dynamics of genes associated with pluripotency (OCT4, NANOG, and REX1) and pancreatic differentiation (PDX1, INS, GCG, and SST). Total RNA of the human pancreas was used as a control for gene expressions. Scale bars: 100 μm.
To investigate the physiological status of the insulin secretion signaling pathways in these spheroid-forming cells, the effects of several insulin agonists (carbachol, tolbutamide, and IBMX) and antagonists (nifedipine and diazoxide) on insulin secretion were examined in the presence of low (3.3 mM) or high (25 mM) glucose concentrations. In all treatment groups, insulin secretion of DTZ+ spheroids was significantly increased in the presence of high glucose compared to low glucose (Fig. 4f). Undifferentiated hESCs did not show the glucose-stimulated insulin secretion. Furthermore, carbachol (a muscarinic cholinergic receptor agonist), tolbutamide (a sulfonylurea inhibitor of ATP-dependent K+ channels), and IBMX (an inhibitor of cyclic AMP phos-phodiesterase) significantly potentiated insulin secretion. In contrast, nifedipine (an L-type Ca2+ channel blocker) and diazoxide (an activator of ATP-dependent K+ channels) inhibited insulin secretion (Fig. 4f).
Gene expression profiles at each stage of differentiation showed that hESC markers OCT4, NANOG, and ZFP42 (REX-1) decreased upon differentiation, but pancreatic endocrine cell-associated genes, PDX1, INS, GCG, and SST, all gradually increased (total RNA of the normal human pancreas were used as positive controls) (Fig. 4g). PDX1 expression was increased during stages I and II, transiently decreased at the end of stage III, but was upregulated again after final maturation at stage IV.
Transplantation of DTZ+ Spheroids Rescues Hyperglycemia in Diabetic Nude Mice
We next characterized the function of islet-like clusters in STZ-induced diabetic nude mice. Monitoring of nonfasting blood glucose levels and body weights showed that the mice that did not receive cells and culture medium (control) or the mice injected with culture medium (sham) exhibited sustained hyperglycemia and diabetic weight loss until they died 30–40 days after STZ injection (Fig. 5a, b). In contrast, transplantation of DTZ+ cell aggregates profoundly decreased glucose levels within 50 days and maintained normal glucose levels. Furthermore, graft removal at 100 days after transplantation resulted in a rapid return of hyperglycemia (Fig. 5a). Consistent with these results, the mice grafted with DTZ+ cell aggregates continued to gain body weight over time (Fig. 5b). To assess the ability of glucose-stimulated insulin release, an IPGTT was performed on the engrafted mice (n = 6) and control healthy mice (n = 4) at day 45 after transplantation. The results revealed that the mice that received DTZ+ cell clusters showed a similar pattern of blood glucose clearance to that of normal control mice (Fig. 5c). Human insulin was also detected in the sera of grafted animals by ELISA, but not in control normal and sham-operated mice (Fig. 5d).

Transplantation of hESC-derived aggregates into STZ-induced diabetic mice. (a, b) Changes in nonfasting blood glucose levels and body weights in control STZ-diabetic mice (n = 4), sham-operated STZ-diabetic mice (n = 4), and STZ-diabetic mice transplanted with hESC-derived aggregates (n = 6) over 110 days after transplantation. *p < 0.05 versus control and sham. (c) Average blood glucose levels in normal mice (control, n = 3) and STZ-diabetic mice grafted with hESC-derived aggregates (n = 3) in response to an IPGTT after 45 days of transplantation. (d) Levels of insulin detected in control diabetic mice, sham-operated diabetic mice, and diabetic mice transplanted with hESC-derived aggregates after 30 days of transplantation. *p < 0.05 versus control and sham. (e–i) Immunofluorescence labeling of grafts with anti-INS, -human nuclei (hNuclei), -pro-INS, -c-pep, -GCG, and -SST antibodies. Scale bars: 100 μm (e); 50 μm (f–i).
Immunohistochemistry of the removed grafts 100 days following transplantation showed that the grafts contained INS+ cells, which were immunoreactive to an antibody against human nuclei in spherical clusters (Fig. 5e). INS+ cells are also positive for pro-INS (Fig. 5f) and c-pep (Fig. 5g), suggesting that these cells actively synthesize and process insulin. GCG+ and SST+ cells were also detected in the clusters (Fig. 5h, i), but cells that coexpressed INS and GCG were not observed in the grafts. Taken together, these results demonstrate that hESC-derived DTZ+ 3D cell clusters continue to exhibit pancreatic features and function in diabetic animals after transplantation.
Discussion
Pancreatic islets are complex microorgans composed of five different types of endocrine cells that show a close topological association in a 3D cellular structure (15). A number of ESC studies have tried to generate functional β-cells by mimicking pancreatic development (13), but many of these studies generated pancreatic cells under 2D adherent culture conditions during the final maturation stage of their differentiation protocols. In this study, we developed a four-stage procedure for differentiating hESCs into islet-like 3D spheroids by modifying and optimizing our previous method (43).
Appropriate culture conditions are critical for the development of efficient methods for generating functional pancreatic endocrine cells in vitro. Although a similar spectrum of differentiation factors, including Wnt, activin, and RA, has been used to initiate β-cell differentiation in hESCs, their combination and concentrations, treatment window and duration, and sequence of the treatments differ, depending on the protocol (13,16). In stage I, we observed that a transient upregulation of T and MixL1 was followed by the increased expression of endodermal genes, including FOXA2, SOX17, and CXCR4 after sequential treatment with Wnt3a, activin A, and noggin. Subsequently, the addition of RA at the end of stage I highly upregulated PDX1 expression. These data suggest that the sequential conversion of hESCs into mesendoderm, endoderm, and pancreatic endoderm was successfully achieved using our protocol as in normal pancreatic development.
In normal pancreatic development, early endocrine progenitor cells form aggregates that develop into pancreatic islets as they migrate through the surrounding mesenchyme (9). Signaling molecules secreted from the notochord, dorsal aorta, and mesenchyme play critical roles in aggregate formation, a key event in the development of pancreatic islets (6). In stage II, we discovered that immediately after RA treatment, dissociation of EBs and further differentiation in the presence of bFGF and activin βB produced many cellular aggregates under 2D adherent conditions. Although cyclopamine has been used to block SHH signaling during the pancreatic differentiation of pluripotent stem cells (4,23,37), our data showed that the combination of bFGF and activin βB was more effective in the upregulation of PDX1 expression at the mRNA and protein levels. A previous study suggested that bFGF stimulates clustering of human islet-derived precursor cells and the formation of 3D pancreatic islets in vitro by acting as a paracrine chemoattractant (12). Thus, it is possible that bFGF contributes to the formation of cellular aggregates that express PDX1 in stage II of our protocol. Another important factor that is possibly involved in 3D cell clustering in stage II is B27. Currently, the exact action mechanism of B27 in cluster formation is unclear since B27 is composed of many different hormones, growth factors, and antioxi-dants (2). However, we previously observed that B27 was a critical factor in the generation of spherical clusters that express early hepatic markers after dissociation of Wnt3a/ BMP4-treated hEBs (22). A recent study also showed that both B27 and bFGF markedly promoted the recruitment and aggregation of cells in 3D colonies during the direct lineage conversion from 5-azacytidine-treated human skin fibroblasts into insulin-secreting cells (40).
To our knowledge, this study is the first report showing that blocking a potential inhibitory effect of mesenchymal cells could promote endocrine differentiation of hESC-derived pancreatic progenitor cells. We showed for the first time that both mensenchymal cells and sulfated proteoglycans were preferentially coexisted with PDX1+ cells in 3D clusters during directed differentiation of hESCs into pancreatic cells. We also demonstrated that chlorate, an inhibitor of proteoglycan sulfation, was able to increase the expression of Neurog3 and NeuroD1, both of which are expressed during the maturation of pancreatic endocrine progenitors into β-cells (7). Furthermore, the chlorate-induced upregulation of these two genes was accompanied by the downregulation of PDX1 in stage III, suggesting the proper transition of hESC-derived PDX1+ pancreatic progenitor cells into early endocrine cells. Finally, RT-PCR analyses of all stages demonstrated that PDX1 expression decreased in stage III and was upregulated again after the final stage IV, demonstrating a biphasic pattern of expression similar to that in normal pancreatic development. Taken together, these data suggest that our protocol provides differentiating hESCs with a proper microenvironment that mimics the early and late development of normal pancreatic endocrine cells.
The relationship between β-cells and non-β-cells for proper glucose sensing and insulin release has been controversial in mature islets (19,24,32,35). However, in developing islets, the close intercellular communication between β-cells and non-β-cells in the endocrine architecture may be important for the maturation of immature islets to functional islets. In stage IV, using DTZ, which was previously shown to identify the endocrine pancreas developed from mouse and human ESCs (4,44), we isolated 3D clusters without dissociation and further matured the purified aggregates in suspension for an additional 6 days. Our data showed that the purified aggregates produced pancreatic hormones (i.e., insulin, glucagon, somatostatin, and c-peptide) and released insulin in response to both changes in glucose concentrations and addition of pharmacological drugs in vitro. The percentage of cells that were positive for insulin was 6–7% of total differentiated cells, but increased up to 27% after the purification of DTZ clusters. Considering β-cells constitute about 65–80% of total islet cells in the pancreas, further studies should be followed to increase the differentiation yield and insulin secretion rate.
Transplantation studies demonstrated that grafted isletlike aggregates functioned in STZ-induced diabetic mice. In our previous study, dissociated PDX1+ spherical clusters were unable to survive and failed to differentiate in vitro into insulin-producing cells under 2D culture conditions (43). Thus, it can be speculated that the formation of 3D islet-like cellular aggregates promoted interactions between β-cells and non-β-cells, or between β-cells, improving the survival and function of differentiating hESC-derived β-cells. It will be interesting to investigate whether differentiation into 3D islet-like cellular aggregates has benefits in obtaining and transplanting more functionally mature β-cells compared to 2D conditions during the final differentiation stage of ESCs and iPSCs. No sign of tumor formation was observed in animals over 110 days after transplantation of DTZ+ cell aggregates. However, further long-term in vivo studies are needed to assess the safety of these cells.
Recently, there has been an increase in the number of clinical islet transplantation cases (34). Thus, the generation of 3D islet-like cellular aggregates from human pluripotent stem cells may overcome the primary hurdle with islet transplantation, the shortage of cadaveric donor islets.
Footnotes
Acknowledgments
This work was supported by the Industrial Strategic Technology Development Program (Project No. 10041913, Stem cell culture system based on surface-nano-structure control) funded by the Ministry of Trade, Industry and Energy (MI, Republic of Korea) and by the Bio and Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIP) (2012M3A9B4028636). The authors declare no conflict of interest.