
Abstract
Etk/Bmx (epithelial and endothelial tyrosine kinase, also known as BMX), a member of the Tec (tyrosine kinase expressed in hepatocellular carcinoma) family of protein-tyrosine kinases, is an important regulator of signal transduction for the activation of cell growth, differentiation, and development. We have previously reported that activation of Etk leads to apoptosis in MDA-MB-468 cells. The purpose of this study was to examine the role of Etk in neuronal injury induced by H2O2 or ischemia. Using Western blot analysis and immunohistochemistry, we found that treatment with H2O2 significantly enhanced phosphorylation of Etk and its downstream signaling molecule Stat1 in primary cortical neurons. Inhibiting Etk activity by LFM-A13 or knocking down Etk expression by a specific shRNA increased the survival of primary cortical neurons. Similarly, at 1 day after a 60-min middle cerebral artery occlusion (MCAo) in adult rats, both phosphorylated Etk and Stat1 were coexpressed with apoptotic markers in neurons in the penumbra. Pretreatment with LFM-A13 or an adenoviral vector encoding the kinase deletion mutant EtkΔk attenuated caspase-3 activity and infarct volume in ischemic brain. All together, our data suggest that Etk is activated after neuronal injury. Suppressing Etk activity protects against neurodegeneration in ischemic brain.
Introduction
Cerebral ischemia triggers a complex progression of biochemical and molecular changes that impair neurologic functions. During the first few hours after ischemia, neurons in the ischemic penumbra suffer transiently reversible damage and then ultimately undergo death by apoptosis (17,24). Evidence has shown involvement of signaling molecules and transcription factors in ischemia-induced cell apoptosis (12,13,15,39), including calcium/calmodulin-dependent kinases (CaMKs), mitogen-activated protein kinases (MAPKs), and signal transducers and activators of transcription (Stat1) (16). Reactive oxygen species (ROS) and inflammatory reactions, which are generated during ischemic injury (27), are also important mediators of cerebral injury. ROS activate MAPK cascades and inflammatory cytokines such as interleukin-1 (IL-1), IL-6, and tumor necrosis factor-α (TNF-α), which then activate caspase cascades that lead to cellular apoptosis (26,33). Despite intensive investigations over the past decades, the molecular basis underlying ischemic injury development and progression is not yet completely understood (7,20).
Epithelial and endothelial tyrosine kinase/bone marrow tyrosine kinase gene in chromosome X protein (Etk/Bmx), a member of the Tec (tyrosine kinase expressed in hepatocellular carcinoma) family of nonreceptor tyrosine kinases, is present in epithelial, endothelial, and granulomonocytic cells (23). Etk and its family members have been shown to be major modulators of signaling pathways initiated by various extracellular stimuli, including growth factors, cytokines, hormones, and extracellular matrix (22). Several reports have validated the importance of Etk in differentiation, proliferation, cell motility, and apoptosis (4,5,32,37).
Etk also plays a pivotal role in signal transduction in apoptosis. Etk was reported as a substrate for caspase-3 during apoptosis and the cleavage product of Etk potentiates cell death in the human prostate cancer cell line PC3 under apoptotic stimuli (37). We previously found that activation of Etk by epidermal growth factor (EGF) increases tyrosine phosphorylation of Stat1 in breast cancer cells, leading to apoptosis (4). Additionally, Etk has been implicated in the signaling pathway mediating the hypoxia-induced response in keloid fibroblasts (3). Taken together, these data suggest that Etk is activated after injury in nonneuronal cells. Its role in ischemic brain injury has not yet been reported.
In this study, we found that Etk was phosphorylated in cortical neurons after ischemic insults. Suppressing Etk expression or its activity attenuates H2O2 and ischemia-mediated neural degeneration in vitro and in vivo. Our data suggests that Etk plays an important role in ischemia-induced cell death.
Materials and Methods
Primary Cortical Culture and Lentivirus Infection
Primary cortical neuron cultures were prepared from the cerebral cortex of gestation day 17 embryos from Sprague-Dawley rats. Briefly, pooled cortical tissues were minced by mechanical trituration and treated with trypsin (0.1%, Invitrogen) for 10 min at 37°C. Cells were dissociated by gentle pipetting in minimum Eagle's medium and plated into six-well plates precoated with poly-L-lysine (0.02 g/L) at a density of 2 × 106 cells/well. After 4-h incubation, the culture medium was replaced with Neurobasal medium (Invitrogen) supplemented with N-2 supplement, 200 mM glutamine, 10 mM glutamate, and penicillin/streptomycin. Cells were maintained in 5% CO2 at 37°C. For virus infection, cells at 7 days in vitro were infected with Lenti-shEtk for 90 min in Opti-MEM. Cells were washed with PBS once and fresh N-2 Neurobasal medium once and were then incubated for another 72 h. The lentivirus vectors for Etk shRNA were generous gifts from Dr. Yun Qiu (11). The oligonucleotides encoding Etk shRNA were 5′-TGGAGCTGGGAAGTGGCCAGTTCAAGAGACTGGCCACTTCCCAGCTCCTTTTTTC-3′ and 5′-TCGA GAAAAAAGGAGCTGGGAAGTGGCCAGTCTCTTGAACTGGCCACTTCCCAGCTCCA-3′. These oligonucleotides were annealed and ligated downstream of the U6 promoter. Lentivirus preparation and infection were carried out as described previously (25).
Western Blot
Ischemic tissues and primary cortical neuron cultures were lysed in radioimmunoprecipitation assay (RIPA) buffer. Cell lysates (50–100 μg) were resolved on 8% to 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS/PAGE) gel, and transferred onto nitrocellulose membranes. Subsequently, blots were incubated with antibodies raised against the following proteins: anti-Bmx (Transduction Laboratories), pTyr-Etk (pEtk, Cell Signaling), Stat1, pTyr701-Stat1 (pStat1, Transduction Laboratories), and actin (Sigma). Donkey peroxidase-conjugated anti-rabbit or anti-mouse antibodies (Amersham Pharmacia Biotech) were used and binding was revealed by chemiluminescence (ECL; Amersham Pharmacia Biotech).
Determination of Infarct Size
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of National Defense Medical Center Laboratory Animal Center (NDMCLAC). Adult male Sprague-Dawley rats (weight >300 g) were used for this study. After intracerebral administration of the tyrosine kinase inhibitor LFM-A13 (10 μg/kg) or 25 μl of Ad-EtkΔk (2 × 1010 pfu/ml) using glass micropipettes, animals were subjected to middle cerebral artery occlusion (MCAo) as previously described (34). The injection site was 2.0–3.0 mm below the dura, 0.5–1.5 mm posterior to bregma, and 4.0–4.5 mm lateral to the midline. Systemic parameters including blood gases were within normal range and differences between the control group and the treatment groups were not significant. Animals were killed after 24-h reperfusion, the brains were removed and a series of 2-mm coronal slices were obtained and stained in 2% triphenyltetrazolium chloride (Sigma) in 0.9% saline, then fixed in 4% paraformaldehyde. The infarction area, which was not stained, was measured using a digital scanner as previously described (35).
Recombinant Adenovirus Generation
The recombinant adenovirus expressing EtkAk was constructed and generated according to the manufacturer's instruction in the adenovirus expression vector kit (TaKaRa). In brief, EtkAk cDNA (4) was inserted into the Staphylococcus warneri (SwaI) site of a pAxCAwt cosmid vector, then cotransfected with the complexes of adenovirus genomic DNA and terminal protein into 293 cells to generate virus. After confirmation, the virus was subjected to amplification and titration.
Immunohistochemistry
Twenty-four hours after reperfusion, rats were perfused through the ascending aorta with 100 ml of cold normal saline followed by 100 ml of 4% paraformaldehyde (PFA) in PBS. Brains were removed and postfixed in the same fixative for 3 days followed by 30% sucrose for 1 week. Sections were cut at a thickness of 12 μm in a freezing microtome and stored at −20°C. For immunostaining, tissue sections or primary cortical cells were fixed with 4% PFA for 10 min. After several washes in PBS, they were incubated with blocking buffer containing 0.3% Triton X-100 and 4% bovine serum albumin for 1 h at room temperature, and were then stained with the desired primary antibody reconstituted in PBS, 2% goat serum at 4°C for 14–16 h. Dilutions of the anti-pEtk (Cell Signaling), anti-pStat1, anti-caspase-3, anti-neurofilament M (NF), anti-glial fibrillary acidic protein (GFAP) (Transduction Laboratories) antibodies were 1:100. After three rinses in PBS, sections were incubated with goat anti-rabbit IgG fluorescein isothiocynate FITC conjugate (1:100 Jackson Immunoresearch) and goat anti-mouse IgG Rhodamine conjugate (1:100; Jackson Immunoresearch) for 1 h at room temperature. For terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, sections were incubated with TUNEL reaction mixture (Boehringer Mannhein GmbH) for 1 h at 37°C. 4′,6-Diamidino-2-phenylindole (DAPI; 1 μg/ml) was added to the mixture during the last 15 min. For identifying nonviable neurons, tissue sections were stained with Fluoro-Jade B (FJB, Chemicon) (28) and counterstained with DAPI. After washing in PBS several times, sections were mounted with Crystal Mount (Sigma) and analyzed by a Leica microscope, a SROT RT™ charge-coupled device (CCD) camera (Diagnostic Instruments), or laser-scanning confocal microscope (Bio-Rad, MRC-1000).
MTT Assay
Primary cortical neurons (2 × 105 per well) were seeded in 24-well plates. Cells were treated with 10 μM LFM-A13 or Lenti-shEtk for 2 days. After treatment with H2O2 for 24 h, tetrazolium MTT [3-(4,5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] was added to the medium at a final concentration of 0.5 mg/ml for 1.5 h, followed by dissolving the formazan with 200 μl dimethyl sulfoxide (DMSO). The absorbance of samples at 550 nm was measured using an ELISA plate reader.
Statistics
Data are presented asmean ± SD. One-way ANOVA and post hoc Newman-Keuls tests were used for statistical comparison. A difference was considered to be statistically significant at p < 0.05.
Results
Etk Is Expressed in Primary Cortical Neurons and Activated by H2O2
Immunocytochemical analysis was carried out on day 8 in primary cortical neuronal cultures. Using confocal microscopy, immunoreactivity of Etk was found mainly in NF-positive (Fig. 1A, top panel), but not GFAP-positive (Fig. 1A, middle panel) cells. Treatment with 100 μM H2O2 enhanced immunoreactivity of phosphorylated Etk in NF-positive neurons (Fig. 1A, lower panel). There was a time-dependent increase in pEtk and pStat1 immunoreactivity after exposure to H2O2 as examined by Western analysis (Fig. 1B). Activity of pEtK and pStat1, normalized to actin, was significantly enhanced at 30 and 60 min after H2O2 exposure (*p < 0.05, one-way ANOVA, post hoc Newman-Keuls test) (Fig. 1C). Cell survival was indirectly measured by MTT assay (Fig. 2). We found that H2O2 (100 μM for 24 h) significantly reduced MTT activity. Pretreatment with LFM-A13 (10 × 10–3 μM) or Lenti-shEtk (MOI = 2) significantly antagonized H2O2-mediated cell death [*p < 0.05, F(3, 53) = 124.228, one-way ANOVA] (Fig. 2).

Epithelial and endothelial tyrosine kinase (Etk) is expressed in primary cortical neurons and is activated by H2O2. (A) Immunostaining of Etk is shown in primary cortical neurons. Etk-positive cells are colocalized with neurofilament (NF)-positive neurons but not glial fibrilary acidic protein (GFAP)-positive cells. Scale bar: 80 μm. Tyrosine phosphorylation of Etk is activated and colocalized with NF-positive neurons after H2O2 treatment for 60 min. (B) Western blot analysis of primary cortical neurons after H2O2 treatment. Levels of pEtk and pStat1 (phosphorylated signal transducers and activators of transcription 1) are increased after H2O2 treatment. (C) Results of H2O2 treatment using densitometric analysis. Etk and Stat1 phosphorylation, normalized to actin, are enhanced at 30 and 60 min. *p < 0.05, one-way ANOVA.
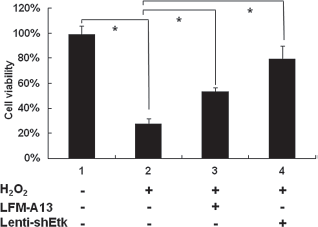
Both LFM-A13 and Lenti-shEtk reduce H2O2-induced cell death in primary cortical neurons. Primary cortical neurons were treated with either 10 μM LFM-A13 or Lenti-shEtk [multiplicity of infection (MOI) = 2] for 2 days then cultured in 100 μM H2O2 for 24 h. Cell viability was measured by 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells with LFM-A13 or Lenti-shEtk treatment showed increased viability when compared with cells treated with H2O2 alone. Data shown are the mean ± SD values from a representative experiment, performed in triplicate, which was repeated four times with similar results. *p < 0.05, one-way ANOVA compared to H2O2 added alone.
Activation of Etk and Stat1 in Ischemic Brain
A total of six stroke rats and six controls were used to examine Etk and pEtk expression in vivo. As shown in Figure 3A and B, pEtk level in the ischemic cortex was significantly increased from 15 min to 24 h after 1-h MCAo (p < 0.05, one-way ANOVA). Similarly, phosphorylation of Stat1 Tyr701 also significantly increased at 2 and 24 h after MCAo (*p < 0.05, one-way ANOVA) (Fig. 3C, D).

Activation of Etk and Stat1 after cerebral ischemia. Western blot analysis of Tyr phosphorylation of Etk (A) and Stat1 (C) in the ischemic cortex 1 h after middle cerebral artery (MCA) ligation and different times of reperfusion. (B, D) The densitometric measurements show increased activation of Etk and Stat1 after 15 min of reperfusion. Levels of actin represent an internal control. Results are representative of three independent experiments. *p < 0.05, one-way ANOVA compared with control.
The expression and distribution of pEtk were examined in nine rats using immunohistochemistry. There was a time-dependent increase of pEtk immunoreactivity in the lesioned cortex from 8 to 72 h after MCAo (Fig. 4A). Minimal pEtk activity was found in the contralateral cortex (Fig. 4A). Using confocal microscopy, we found that pEtk was coexpressed with NeuN, NF, or FJB in neurons in the ischemic penumbra at 24 h poststroke (Fig. 4B). The association of Etk with apoptosis is supported by the observation of the presence of pEtk activity in TUNEL-positive cells and caspase 3-positive cells (Fig. 4C, D). Similar to pEtk, pStat1 immunoreactivity was found in the lesioned cortex at 24 h poststroke (Fig. 4E). These pStat1-positive cells were also coexpressed with FJB (Fig. 4F).
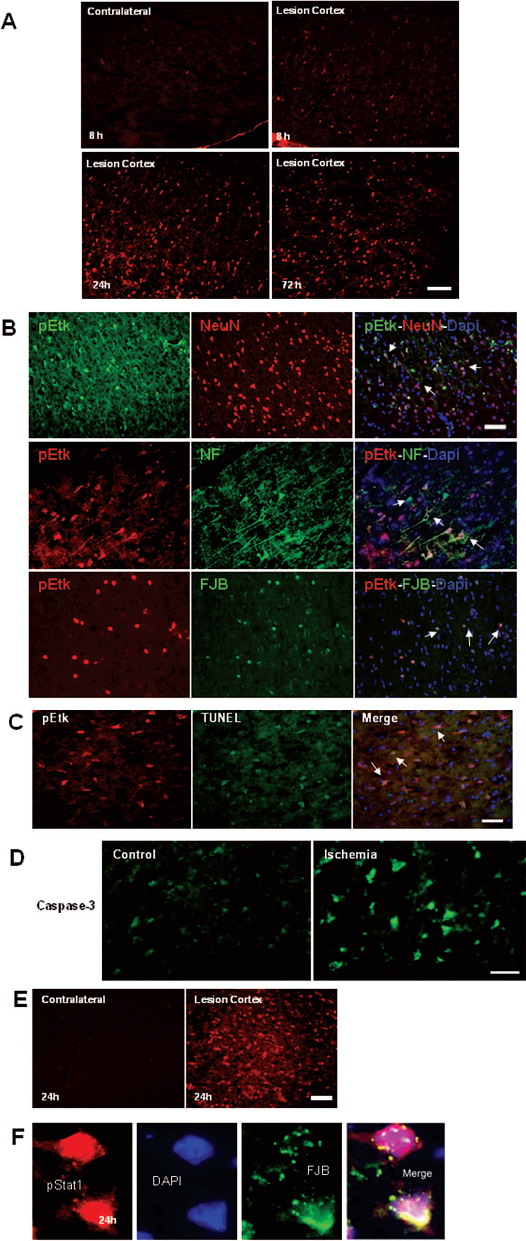
Immunohistochemial study of Etk and Stat1 activation after 1-h MCAo and different times of reperfusion. (A) Immunoreactivity of Tyr phosphorylated Etk (red) increased in the lesioned cortex in a time-dependent manner. (B) After 24-h reperfusion, pEtk is colocalized with NeuN-positive, NF-positive, or FJB-positive neurons within the ischemic cortex (arrows). (C) pEtk immunoreactivity (red) and TUNEL-positive nuclei (green) are colocalized (arrows) in lesioned cortex 24 h after reperfusion. (D) Caspase-3 (green) is also induced in lesioned cortex 24 h after reperfusion. (E) Stat1 is activated in lesioned cortex after 24 h reperfusion (red). (F) Confocal images of pStat1-positive neurons (red) and FJB-positive (green) cells, which are colocalized within the lesioned cortex 24 h after reperfusion. These findings suggesting a link between Etk activation and ischemia-induced neuronal apoptosis. Scale bar: 40 μm.
Neural Protection by Suppressing Etk Activity in Ischemic Brain
Adult rats were treated with Etk kinase inhibitor LFM-A13 (n = 9) or vehicle (n = 9), as well as mutant Ad-EkAk (n=9) or Ad-green fluorescent protein (GFP) (n = 9) given intracerebrally, at 15 min or 7 days, respectively, before MCAo. Since vehicle and Ad-GFP did not alter the size of infarction, data from animals receiving these treatments were combined (Fig. 5).
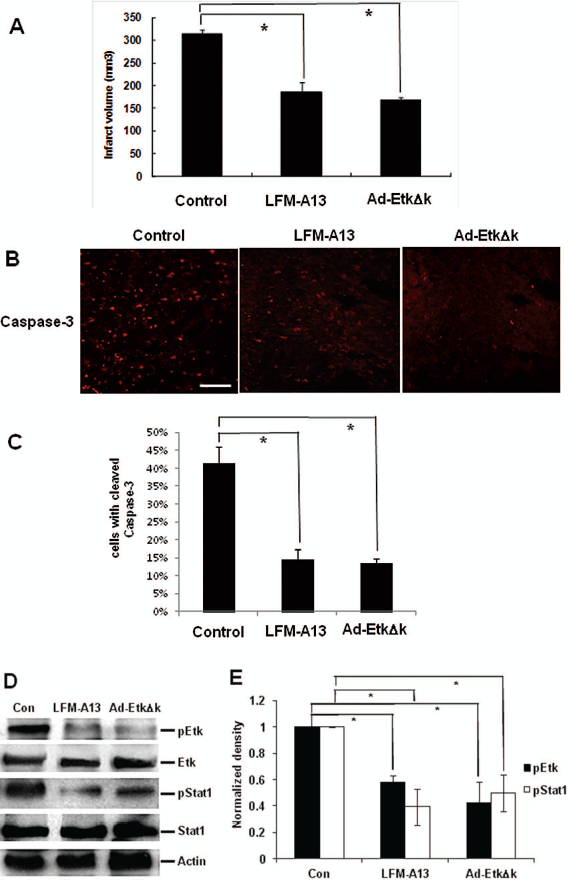
The effects of LFM-A13 and Ad-EtkΔk on ischemic brain injury. (A) Twenty-four hours after reperfusion, infarct volume is significantly reduced in LFM -A13- or Ad-EtkΔk-treated rats compared with control animals (n = 9 per group). Data represent the mean ± SD. *p < 0.05, on- way ANOVA compared with control. (B) Ischemia-induced activation of caspase-3 (red) was reduced in the cerebral cortex of LFM-A13- or Ad-EtkΔk-treated rats. Scale bar: 100 μm. (C) Bar graphs indicate quantitative analysis of cleaved caspase-3-positive cell counts 24 h after reperfusion. (D) Expression of pEtk and pStat1 was decreased after LFM-A13 or Ad-EtkΔk treatment in the cerebral cortex. (E) Densitometric measurements show inhibition of phosphorylation by LFM-A13 and Ad-EtkΔk. Levels of actin represent an internal control. Results are representative of three independent experiments. *p < 0.05, one-way ANOVA compared with control.
Pretreatment with LFM-A13 or mutant Ad-EkAk significantly reduced infarct volume (*p < 0.05, one-way ANOVA)(Fig. 5A) and the density of cleaved caspase-3-positive cells in the ischemic cortex (*p < 0.05, one-way ANOVA) (Fig. 5B, C) at 24 h postreperfusion. In addition, pEtk and pStat1 protein levels were significantly reduced by LFM-A13 or Ad-EkAk in stroke brain, suggesting EtK activity was suppressed after these treatments. In contrast, EtK and Stat1 level were not altered because the kinase deletion mutant Ad-EkΔk only suppresses phosphorylation of Etk but not its expression (Fig. 5D, E).
Discussion
Etk is important in the signal transduction pathway for EGF and vascular endothelial growth factor (VEGF), in epithelial cell apoptosis, and in endothelial angiogenesis. In addition, activation of Etk in hydrogen peroxide-treated human umbilical vein endothelial cells (HUVECs) suggests a correlation between Etk and ischemic damage. In this report, we utilized the MCAo animal model to study the role of Etk. This is the first report illustrating the activation of Etk and Stat1 after free radical or ischemic brain injury.
Association of Etk with brain injury was first demonstrated here in primary cortical neurons. Phosphorylation of Etk is associated with various kinases and is involved in diverse signaling pathways (22). H2O2 increased phosphorylation of Etk and Stat1 with respect to the duration of treatment. Furthermore, suppression of EtK phosphorylation by pretreatment with either the Etk inhibitor LFM-A13 or Lenti-shEtk attenuated H2O2-mediated cell death. These data suggest that phosphorylation of Etk is one of the important mediators for cell death after free-radical damage. Inhibiting Etk phosphorylation enhances cell survival after such injury in vitro.
The role of Etk in ischemic injury was also seen here in the in vivo studies. The cerebral cortex of rats undergoing MCAo showed increased phosphorylation of Etk and Stat1 by Western blot and immunofluorescence. Furthermore, cerebral cortical infarction volume and caspase-3 activity were suppressed in stroke rats pretreated with LFM-A13 or Ad-EtkΔk. These data suggest that Etk activity is directly associated with neural injury in the ischemic MCAo rat model.
Inhibition of apoptosis by Etk/Bmx tyrosine kinase may be cell specific. Etk was first reported to inhibit apoptosis in prostate cancer (LNCaP cells) in 1999 (38), and was later found to exhibit bidirectional inhibition of p53 transcriptional activity and regulate apoptotic effects in prostate cancer cells (11,38). In small cell lung cancer cells, upregulation of tyrosine kinase Etk may be a mechanism involved in protection from apoptosis; interestingly, Bcl-2 and Bcl-X(L), but not p53, contribute to doxorubicin-induced apoptosis through the Etk pathway (9). For intrahepatic cholangiocarcinoma (ICC), Etk/Bmx may be involved in the development of ICC and is a predictor of poor prognosis for ICC. The same study showed that hepatitis C virus (HCV) infection and NF-κB was unrelated to Etk/Bmx expression (10). Intriguingly, in MDA-MB-468 breast cancer cells, activation of Etk is essential for transduction of EGF-induced apoptotic signaling (4).
Ischemic brain injury can induce neural degeneration through inflammation (16,29) and apoptosis (6,8,14). Overactivation of N-methyl D-aspartate (NMDA) receptors during cerebral ischemia results in increased formation of ROS, particularly nitric oxide (NO) (1). Previous studies have shown that phosphorylation of EtK is associated with inflammation (18,19) and free radical generation in heart (40). In our study, we found that ischemia enhanced Etk phosphorylation while suppression of this reaction reduced neurodegeneration in stroke brain. Further studies are needed to determine whether activation of inflammation is mediated through Etk phosphorylation in stroke brain.
Previous studies have indicated that Stat1 participates in ischemia-induced cell death (21,30). Stat1 can be activated by oxidative stress and ischemia (2,31). Knocking out Stat1 reduced ischemic brain damage (36). Our data also indicate that Stat1 was phosphorylated in tissue culture or brain cells after free radical or ischemic injury. Suppression of EtK activity by pharmacological agents or genetic manipulation attenuated Stat1 phosphorylation and induced protection. These results support the view that EtK promotes cell death through Stat1 activation in stroke brain. In addition to Stat1, other members of the Stat family are also expressed in the brain. Their roles in ischemic brain damage remain to be elucidated.
In conclusion, the present data provide the first demonstration that Etk is expressed in neurons and can be activated through phosphorylation after cerebral ischemia. Suppressing EtK activity protects against neurodegeneration in ischemic brain.
Footnotes
Acknowledgments
This study was supported by the National Health Research Institutes Postdoctoral Fellowship Award PD9404, and National Science Council Grant NSC95-2314-B016-026, NSC-95-3112-B016-002, and NSC97-2314-B038-007. The authors declare no conflict of interest.