
Abstract
Bone marrow mononuclear cells (BMMNCs) can be used to treat patients with myocardial infarction, since BMMNCs can differentiate in vitro toward cardiomyogenic lineages when treated with transforming growth factor-β1 (TGF-β1). However, the in vitro cardiomyogenic differentiation culture process is costly and laborious, and the patients should wait during the culture period. In this study, we hypothesize that BMMNCs implanted in cardiomyogenically undifferentiated state to myocardial infarction site would differentiate cardiomyogenically in situ when exogenous TGF-β1 is delivered to the cell implantation site. Heparin-conjugated poly(lactic-co-glycolic acid) nanospheres (HCPNs) suspended in fibrin gel were used as a TGF-β1 delivery system. BMMNCs were labeled with a green fluorescent dye (PKH67) and implanted into the infarction border zone of rat myocardium using fibrin gel containing HCPNs and TGF-β1. BMMNC implantation using fibrin gel and HCPNs without TGF-β1 served as a control. Four weeks after implantation, the expression of cardiomyogenic marker proteins by the implanted BMMNCs was dramatically greater in the TGF-β1 delivery group than in the control group. This method can significantly improve the stem cell therapy technology for myocardial regeneration, since it can remove in vitro cell culture step for cardiomyogenic differentiation prior to cell implantation.
Keywords
Introduction
Implantation of bone marrow mononuclear cells (BMMNCs) to infarcted myocardium can enhance angiogenesis and/or improve cardiac function in animals (9,16,19) and humans (22–25). BMMNCs contain stem cells that can differentiate into various kinds of cell lineages, such as osteogenic, adipogenic, chondrogenic, and myogenic cells (17). Importantly, implanting cardiomyocytes differentiated from bone marrow stem cells results in better myocardial regeneration and greater heart functional improvement than implanting undifferentiated bone marrow stem cells (10). Several methods have been developed to differentiate bone marrow cells into cardiac lineage cells. These methods include coculturing bone marrow cells with cardiomyocytes (4) and treatment with 5-azacytidine (14) or transforming growth factor-β1 (TGF-β1) (10). However, the in vitro cardiomyogenic differentiation process requires an additional culture, which is costly and laborious, and patients with myocardiac infarction should wait during the culture period.
In this study, we tested the hypothesis that BMMNCs that are implanted in cardiomyogenically undifferentiated state to myocardial infarction site would differentiate cardiomyogenically in situ when exogenous TGF-β1 is delivered to the cell implantation site. In situ cardiomyogenic differentiation of implanted BMMNCs can remove the in vitro culture step for cardiomyogenic differentiation prior to cell implantation and could significantly improve the stem cell therapy for myocardial infarction. Previously, heparin-conjugated poly (lactic-co-glycolic acid) (PLGA) nanospheres (HCPNs) suspended in fibrin gel have shown to be able to release basic fibroblast growth factor, a heparin-binding growth factor, in a sustainable manner (8). In the present study, HCPNs suspended in fibrin gel were utilized to deliver TGF-β1, another heparin-binding growth factor (7), locally to BMMNC implantation site in infarcted myocardium in rats for a long period. BMMNC implantation using fibrin gel and HCPNs without TGF-β1 served as a control. Four weeks after implantation, cardiomyogenic differentiation of the implanted BMMNCs that were labeled with a green fluorescent dye (PKH67) prior to implantation was examined immunohistochemically.
Materials and Methods
BMMNC Isolation and Labeling
Bone marrow cells were flushed from the femurs and tibias of Sprague-Dawley rats (male, 200–250 g, Jungang Laboratory Animal, Seoul, Korea) into Dulbecco's modified Eagle's medium (DMEM, Gibco BRL, Gaithersburg, MD, USA). The cell suspension was loaded on Ficoll-Paque density gradient (specific gravity = 1.077, Amersham Biosciences, Arlington Heights, IL, USA), and centrifuged at 2000 rpm for 30 min. BMMNCs were isolated from the layer between the Ficoll-Paque reagent and blood plasma, and washed three times in phosphate-buffered saline (PBS; Sigma, St. Louis, MO, USA). Prior to implantation, BMMNCs were labeled with PKH67 green fluorescent dye (Sigma) for cell membrane labeling. BMMNCs (2 times; 107 cells) were suspended in 1 ml of Diluent C, immediately mixed with 1 ml of PKH67 dye (5), and incubated at room temperature for 3 min. Two milliliters of fetal bovine serum (FBS, Gibco BRL) was added to stop the staining reaction. The mixture was diluted with an equal volume of culture medium [DMEM supplemented with 10% (v/v) FBS and 1% penicillin and streptomycin] and centrifuged at 1200 rpm for 10 min. Finally, PKH67-stained BMMNCs were washed three times with the medium.
Preparation of Heparin-Conjugated PLGA Nanospheres and Fibrin
Biodegradable PLGA nanospheres were prepared using the oil/water emulsion and solvent evaporation-extraction method, as previously described (7). Heparin (Mw 17,000, activity 170 USP units/mg, Sigma) was covalently conjugated to the surface of amino-terminated PLGA nanospheres using a procedure employing standard carbodiimide chemistry (7). Heparin (100 mg) was dissolved in a buffer solution (100 ml, pH 6) of 2-morpholinoethanesulfonic acid (0.05 M; Sigma) containing 0.5 M NaCl. N-Hydroxysuccinimide (0.06 M; Sigma) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (0.12 M; Sigma) were added to the solution to activate the carboxylic acid groups of the heparin. After 4 h of reaction at 4°C, the solution was filtered through a 0.45-μm filter to remove aggregates and byproducts. After amino-terminated PLGA nanospheres were immersed in the reaction solution overnight at 4°C, the HCPNs were washed five times with sonication in distilled water to remove unreacted materials and heparin, and then lyophilized for 2 days. The morphological examination of the HCPNs was performed using a scanning electron microscope (S-4800 UHR FE-SEM, Hitachi, Tokyo, Japan). The size distribution of HCPNs was determined using dynamic laser light scattering (Zetasizer 3000HS, Malvern, UK). The amount of heparin conjugated to the nanosphere surface was determined with the toluidine blue method (8). To load TGF-β1 on the nanosphere, HCPNs (14 mg/ml) and TGF-β1 (2 μg/ml, R&D Systems, Minneapolis, MN, USA) were mixed in 2 ml of PBS. After 30 min, TGF-β1-loaded HCPNs were collected by ultracentrifugation at 14,000 rpm at −4°C for 10 min. Fibrin matrix was purchased from a commercially available fibrin gel kit (Greenplast®, GreencrossPD Co., Yongin, Korea). Plasminogen-free fibrinogen (100 mg) and fibrin-stabilizing factor XIII (44–88 U) were dissolved in 1 ml of plasmin inhibitor aprotinin solution (1000 KIU/ml). Thrombin (400–600 IU) was dissolved in 1 ml of calcium chloride solution (4.9–11.1 mg/ml) for thrombin solution. The fibrinogen and thrombin solutions were mixed at a 1:1 volume ratio to form the fibrin gel.
In Vitro TGF-β1 Release Kinetics
The kinetics of TGF-β1 release from delivery systems was determined with an enzyme-linked immunosorption assay (ELISA). Each delivery system containing TGF-β1 (n = 3 per group) was immerged in a 2 ml microcentrifuge tube containing 1 ml PBS, and the tubes were incubated at 37°C with continuous agitation. At various time points, the supernatant was collected, and the microcentrifuge tubes were replenished with fresh buffer. The amounts of TGF-β1 in the supernatants were determined with an ELISA kit (Human TGF-β1 Duoset®, R&D Systems).
In Vitro TGF-β1 Bioactivity
Rat bone marrow stromal cells (rBMSCs, 1 times; 104 cells/well) were cultured for 14 days on each well of six-well tissue culture plates (Corning Inc., New York, NY, USA) and HCPNs suspended in fibrin gel and loaded with TGF-β1 were fixed on an insert (Transwell®, Corning Inc., New York, NY, USA) in the culture plates. The medium was DMEM containing 10% (v/v) FBS (Gibco) and 1% penicillin/streptomycin. The bioactivity of released TGF-β1 was evaluated with imunocytochemistry, reverse transcriptase polymerase chain reaction (RT-PCR), and Western blot analysis. After 14 days, the cells were immunofluorescently stained with anti-α-myosin heavy chain (α-MHC, Abcam, Cambridge, UK) and anti-α-sarcomeric actin (Abcam), respectively. The staining signals for α-MHC and α-sarcomeric actin were visualized with fluorescein-isothio-cyanate (FITC)-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). The cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA, USA) and examined using a fluorescence microscope (Nikon TE2000, Tokyo, Japan). Synthesized cDNA was amplified by PCR using rat-specific primers. PCR was carried out for 35 cycles of denaturing (94°C, 30 s), annealing (58°C, 45 s), and extension (72°C, 45 s) with a final extension at 72°C for 10 min. As a positive control, TGF-β1 was added daily to the rBMSC culture for 14 days at the same concentration as that of TGF-β1 released from HCPNs suspended in fibrin gel. rBMSC culture with HCPNs suspended in fibrin gel without TGF-β1 served as a negative control. β-Actin served as a housekeeping gene. Fourteen days after cardiomyogenic differentiation treatments, rBMSCs were washed three times with PBS, lysed by adding sodium dodecyl sulfate (SDS) sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mM dithiothreitol, 0.1% bromophenol blue), and scraped. Proteins were electrophoretically separated on 4–10% SDS-polyacrylamide gels and transferred to membrane (Millipore, Bedford, MA, USA). To detect protein, membranes were incubated in primary antibody against α-MHC (Santacruz, Santacruz, CA, USA), α-sarcomeric actin (Santacruz), and α-cardiac actin (Santacruz) overnight at 4°C, then washed and incubated with secondary antibodies conjugated to horseradish peroxidase (Sigma) for 50 min at room temperature. Blots were developed using enhanced chemiluminescence (LumiGLO, KPL Europe, Guildford, UK) as recommended by the manufacturer.
Rat Myocardial Infarction Model
Myocardial infarction was induced in rat myocardium (12). Sprague-Dawley rats (female, 300–350 g, Jungang Laboratory Animal, Seoul, Korea) were anesthetized with an intramuscular administration of ketamine hydrochloride (100 mg/kg) and xylazine hydrochloride (2.5 mg/kg). After general anesthesia and tracheal intubation with 18-gauge intravenous catheters, rats were artificially ventilated with room air (model 683, Harvard Apparatus, South Natick, MA, USA) at 80 breaths per minute. A left thoracotomy was performed on each rat in the fourth intercostals space, the left coronary artery was occluded within the myocardium between the left artery appendage and right ventricular outflow tract using a curved needle and 6-0 silk, the chest was closed in layers, and the pneumothorax was evacuated. After 45 min, the suture that occluded the left coronary artery was removed. All care and handling of animals were performed according to the US National Institute of Health's guidelines for the care and use of laboratory animals.
Cell Implantation
One week after inducing the myocardial infarction, the rats were randomly assigned to one of two treatment groups (n = 5 per group). Rats were anesthetized by an intramuscular administration of ketamine hydrochloride (100 mg/kg) and xylazine hydrochloride (2.5 mg/kg). The anesthetized rats were intubated and placed on a ventilator, prior to administration of the intramyocardial injection of study treatment. BMMNCs (2.0 times; 107 cells/rat) were suspended in fibrin gel (100 μl) containing HCPNs and TGF-β1 (2 ng) and injected to intramyocardial sites bordering the infarcted myocardium (cell + TGF-β1 group). As a control, BMMNCs (2.0 times; 107 cells/rat) were suspended in fibrin gel (100 μl) containing HCPNs and injected to intramyocardial sites bordering the infarcted myocardium (cell only group). Cyclosporine-A (10 mg/kg body weight, CIPOL®, Chong Kun Dang, Seoul, Korea) was administered 2 days before and then daily after cell implantation for the duration of the experiment to immunosuppress the rats.
Histology and Immunohistochemistry
Four weeks after the implantation, the whole hearts were retrieved and fixed in 4% (v/v) paraformaldehyde solution for 24 h, washed PBS for 5 h, and then immersed in 10% (v/v) sucrose solution for overnight. The whole hearts were excised and transversely sectioned across the infarct territory into two blocks. The half tissue blocks were dehydrated with a graded ethanol series, and embedded in paraffin. The specimens at the implantation sites were cut into 4-μm-thick sections and stained with Masson's trichrome method. Fibrosis area was measured as the area of stained fibrotic tissue by Masson's trichrome staining as previously described (10), and the images were analyzed in a blinded fashion with image analysis system (KS400, Zeiss, Munich, Germany) coupled to a light microscope. The fibrosis area was expressed as the percentage of fibrosis area in total cross-sectional area. The half tissue blocks were frozen in liquid nitrogen with Tissue-Tek O.C.T. compound (Miles Laboratories, Naperville, IL, USA) and stored at −70°C. For immunohistochemical staining, 10-μm-thick sections were stained using antibodies against cardiac-MHC (Abcam, Cambridge, UK), connexin 43 (Abcam), and α-sarcomeric actin (Abcam). The sections were counterstained with DAPI and examined using a fluorescence microscope (Nikon TE2000, Tokyo, Japan). The quantitative of engraftment of BMMNCs was expressed as PKH67-positive cells in total cross-sectional area.
Evaluation of Cardiac Functions
To evaluate cardiac functions of rats (n = 3 per group) with myocardiac infarction that were treated with either PBS injection, BMMSCs injection, or BMMSC + TGF-β1 injection, left ventricular catheterization was performed at 14 days after treatment. Millar Micro-tip 2 F pressure-volume transducer (model SPR-838, Millar Instruments, Houston, TX, USA) was introduced into the left ventricle via the right common carotid artery under zoletil (20 mg/kg) and xylazine (5 mg/kg) anesthesia. Real-time pressure-volume loops were recorded and all data were analyzed with PVAN 3.6 software (Millar Instruments).
RT-PCR Analysis
RT-PCR was performed 4 weeks after implantation. Total RNA was isolated from retrieved heart using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Specimens were lysed and homogenized with 1 ml of TRIzol reagent, added to 200 μl of chloroform, and centrifuged at 12,000 rpm for 15 min. The RNA pellets were washed with 75% (v/v) ethanol, air-dried, and dissolved in RNase-free water. Reverse transcription was performed with 5 μg of pure RNA using SuperScript™ II reverse transcriptase (Invitrogen). Osteoblasts were isolated from calvaria of Lewis rats (SLC, Tokyo, Japan) by a digestive enzymatic process. The calvaria were isolated, and all connective tissues were removed carefully. The parietal bones were minced into pieces using sterile surgical scissors. Osteoblasts were isolated using an enzyme solution containing type 1 collagenase (1.37 mg/ml; Sigma) and trypsin (0.5 mg/ml; Sigma). Following 30-min incubation, the released cells were discarded to prevent contamination with other types of cells (e.g., fibroblasts). The minced bones were redigested with the enzyme solution for 30 min, and the supernatant was transferred to the culture medium, which consisted of DMEM (Gibco BRL) containing 10% (v/v) FBS (Gibco BRL), 1% (v/v) penicillin-streptomycin (Gibco BRL), 10 mM β-glycerophosphate (Sigma), 50 mg/ml L-ascorbic acid (Sigma), and 100 nM dexamethasone (Sigma). This process was repeated three times, and, finally, the collected solution was centrifuged for 10 min at 1500 rpm. Cells were plated into tissue culture flasks and cultured in a humidified incubator at 37°C with 5% (v/v) CO2. Synthesized cDNA was amplified by PCR using rat-specific primers shown in Table 1. PCR was carried out for 30 cycles of denaturing (94°C, 30 s), annealing (58°C, 45 s), and extension (72°C, 45 s) with a final extension at 72°C for 10 min. PCR products were visualized by electrophoresis on 1.5% (w/v) agarose gels with ethidium bromide staining and analyzed with a gel documentation system (Gel Doc 1000, Bio-Rad Laboratories, Hercules, CA, USA). β-Actin served as a housekeeping gene.
Nucleotide Sequences of Rat-Specific Primer Sets for RT-PCR Assays

Statistical Analysis
All quantitative data were expressed as the mean ± SD. Statistical analysis was performed using unpaired Student's t-test and Bonferroni correction where appropriate (InStat, GraphPad Software Inc., San Diego, CA, USA). A value of p < 0.05 was considered statistically significant.
Results
Preparation of Heparin-Conjugated PLGA Nanospheres
Amino-terminated PLGA polymer has been characterized in our previous report (7). A scanning electron microphotograph of the fabricated HCPNs showed that the nanospheres were spherical and discrete particles without aggregation (Fig. 1A) with a diameter of less than 300 nm (Fig. 1B). The amount of heparin conjugated to the PLGA nanospheres was 8 mg/g PLGA nanospheres.
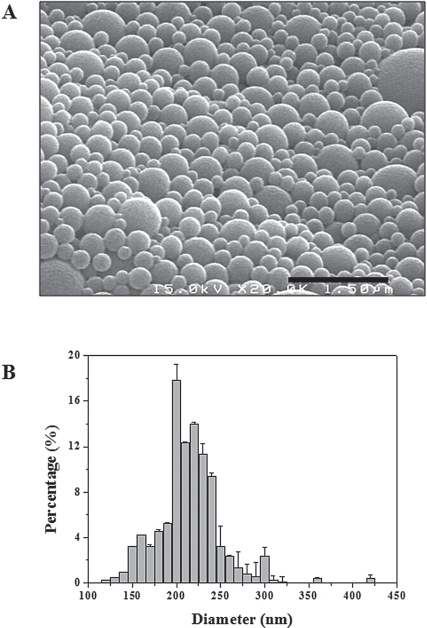
(A) A Scanning electron micrograph and (B) size distribution of heparin-conjugated poly(-lactic-co-glycolic acid) nanospheres (HCPNs). Scale bar: 1.5 μm.
In Vitro TGF-β1 Release Kinetics
The in vitro TGF-β1 release kinetics of three types of delivery systems (fibrin, HCPN, HCPN + fibrin) was compared. The initial burst release and release period dramatically differed depending on the type of delivery system (Fig. 2). The initial release for the first 5 days from fibrin and HCPN was 78.3 ± 2.3% and 46.2 ± 2.3%, respectively, while that from fibrin + HCPN was only 36.4 ± 2.3%. HCPNs released 93.1 ± 1.1% of the loaded TGF-β 1 for 24 days, whereas fibrin released 86.4 ± 2.9% for 7 days. TGF-β1 release from HCPNs was further sustained by suspending HCPNs in fibrin gel.

Profiles of transforming growth factor (TGF)-β1 released from fibrin gel (open box), HCPNs (filled circle), and HCPN suspended in fibrin gel (filled triangle) (n = 5 for each group).
Bioactivity of TGF-β1 Released From Delivery System
To determine whether TGF-β1 released from the delivery system retains its bioactivity, rBMSCs were cultured in a medium containing either HCPNs, TGF-β1-loaded HCPNs suspended in fibrin, or TGF-β1 added daily to the medium. When cultured with medium containing no TGF-β1, rBMSCs did not express cardiomyogenic markers including α-MHC, α-sarcomeric actin, and α-cardiac actin, as evaluated with immunocytochemistry, RT-PCR analysis, and Western blot analysis (Fig. 3). When TGF-β1 was added daily to the culture medium, the cells expressed the cardiomyogenic markers. Importantly, TGF-β1 delivery using HCPNs suspended in fibrin gel also induced expression of the cardiomyogenic markers by rBMSCs, indicating that the released TGF-β1 was bioactive.

The bioactivity of TGF-β1 released from the delivery system. (A) RT-PCR analysis and (B) Western blot analysis to evaluate cardiac-specific gene expression in rat bone marrow stromal cells (rBMSCs) cultured for 14 days with either no TGF-β1 (No TGF-β1), TGF-β1 delivery using HCPNs suspended in fibrin (TGF-β1 delivery), or TGF-β1 daily added into the medium (Daily addition of TGF-β1 to medium).
In Situ Cardiomyogenic Differentiation of Implanted BMMNCs
Masson's trichrome staining of myocardium confirmed that infarction was induced by coronary artery ligation (Fig. 4). Fibrosis occurred in the infarction area indicated as blue color. The fibrosis area in the infarction area was not significantly different between BMMNC and BMMNC + TGF-β1 group (Fig. 4E). BMMNCs labeled with a green fluorescent dye, PKH67, and implanted into myocardial infarction sites were found in the implantation sites 4 weeks after implantation (Figs. 5–7). TGF-β1 delivery did not affect the survival of the implanted BMMNCs at 4 weeks after implantation (Fig. 4F). Immunostaining for cardiac MHC (Fig. 5), connexin-43 (Fig. 6), and α-sarcomeric actin (Fig. 7) merged with PKH67 indicated that cardiomyogenic gene expression by the implanted BMMNCs was far greater in the TGF-β1 delivery group than in the BMMNC group. RT-PCR analysis showed that BMMNCs implanted with TGF-β1 delivery system did not express osteogenic markers such as osteonectin (ON), osteopontin (OP), collagen type I (COL-I), and alkaline phosphatase (ALP) (Fig. 8).

Masson's trichrome staining of rat infracted myocardium 4 weeks after the treatments. (A) Bone marrow mononuclear cell (BMMNC) implantation + TGF-β1 delivery group. (C) BMMNC implantation group. (B) and (D) show infarction sites at a higher magnification. The viable tissues appear red color and fibrous tissues appear blue color. Scale bars (B, D): 100 μm. (E) Fibrosis area of BMMNC implantation + TGF-β 1 delivery group and BMMNC implantation group. (F) The density of PKH67-positive cells (n = 5 animals for each group).

Immunohistochemical staining for cardiac myosin heavy chain (MHC) in the cell implantation region at 4 weeks. (A) BMMNC implantation group. (B) BMMNC implantation + TGF-β1 delivery group. The green color (PKH67) indicates implanted BMMNCs (arrows) and the red color indicates cardiac MHC-positive cells. The yellow color indicates BMMNCs implanted and differentiated into cardiomyocytes (arrow heads). (C) and (D) show a higher magnification of (A) and (B). Scale bars: 100 μm. (E) Percentage of cardiac MHC-positive cells in PKH67–positive cells. There was significant difference between the two groups (n = 5 animals for each group).

Immunohistochemical staining for connexin 43 in the cell implantation region at 4 weeks. (A) BMMNC implantation group. (B) BMMNC implantation + TGF-β1 delivery group. The green color (PKH67) indicates implanted BMMNCs (arrows) and the red color indicates cardiac MHC-positive cells. The yellow color indicates BMMNCs implanted and differentiated into cardiomyocytes (arrow heads). (C) and (D) show a higher magnification of (A) and (B). Scale bars: 100 μm. (E) Percentage of connexin 43-positive cells in PKH67-positive cells. There was significant difference between the two groups (n = 5 animals for each group).
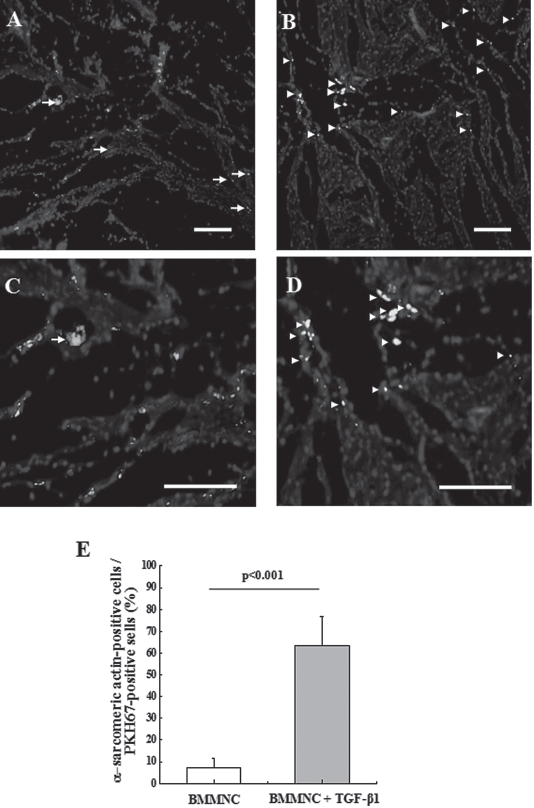
Immunohistochemical staining for α-sarcomeric actin in the cell implantation region at 4 weeks. (A) BMMNC implantation group. (B) BMMNC implantation + TGF-β1 delivery group. The green color (PKH67) indicates implanted BMMNCs (arrows) and the red color indicates cardiac MHC-positive cells. The yellow color indicates BMMNCs implanted and differentiated into cardiomyocytes (arrowheads). (C) and (D) show a higher magnification of (A) and (B). Scale bars: 100 μm. (E) Percentage of α-sarcomeric actin-positive cells in PKH67-positive cells. There was significant difference between the two groups (n = 5 animals for each group).

RT-PCR analysis to evaluate osteogenic gene expression in the cell implantation region at 4 weeks in BMMNC implantation group, BMMNC implantation + TGF-β1 delivery group, and rat carvarial osteoblasts. Expression of osteogenic-specific markers such as osteonectin (ON), osteopontin (OP), collagen type I (COL I), and alkaline phosphatase (ALP) in RT-PCR. RT-PCR analysis was performed using rat-specific primers to detect mRNA expression of osteogenic markers.
Cardiac Function
Treatment with BMMNC + TGF-β1 improved cardiac functions as compared to treatment with BMMNC only (Fig. 9). The myocardiac function was improved in BMMNC + TGF-β1 group compared to BMMNC only group in terms of the volume-related parameters such as ejection fraction and stroke volume as well as the pressure-related parameters such as stroke work.

Myocardiac function of normal rats and rats with myocardiac infarction treated with either PBS injection, BMMNC injection, or BMMNC + TGF-β 1 injection. (A) Ejection fraction. (B) Stroke volume. (C) Stroke work. *p < 0.05 compared to any group except normal group (n = 3 animals for each group).
Discussion
Many studies have reported on regeneration of damaged myocardium by bone marrow stem cell transplantation therapy (10,15,16,19,22,23,25). However, spontaneous cardiomyogenic differentiation of bone marrow stem cells implanted into infarcted myocardium rarely happens (1). A study showed that TGF-β1 induces myogenic differentiation of bone marrow stem cells (10). Previously, we have developed a system for local and sustained delivery of heparin-binding growth factors (e.g., TGF-β1, vascular endothelial growth factor, basic fibroblast growth factor) (8). Here, we demonstrate that local delivery of TGF-β1 using the delivery system to cell implantation site induces in situ cardiomyogenic differentiation of BMMNCs implanted to infarcted myocardium.
The current methods for cardiomyogenic differentiation of bone marrow stem cells have several problems. 5-Azacytidine is a synthetic nucleoside that is commonly used as an inhibitor of DNA methylation. It is a potent inducer of cardiomyogenic differentiation in both embryonic stem cells (23) and bone marrow stem cells (3). However, this synthetic chemical has a possibly harmful, nonspecific demethylating activity (13). Another method for in vitro cardiomyogenic differentiation of stem cells is coculture of stem cells with cardiomyocytes. Coculture with cardiomyocytes induced cardiomyogenic differentiation of BMSCs (4) and adipose-derived mesenchymal stem cells (18). However, the coculture method has a problem that the intimate physical contact between two cell types may lead to fusion of the two different cell types and formation of heterokaryons. Another shortcoming of the coculture method is the difficulty in the separation of cocultured cell populations. The cell separation can be achieved by either fluorescence-activated cell sorting (FACS) (6) or magnetic affinity cell sorting (MACS) (21). However, FACS is skill intensive and requires an expensive instrument. MACS is much cheaper compared to FACS, but the degree of separation purity is much lower.
The important function of a growth factor delivery system would be to deliver growth factors in a bioactive form over a desired period of time. HCPNs were able to release TGF-β1 for 4 weeks, while fibrin released most of the loaded TGF-β1 within 1 week (Fig. 2). The sustained release of TGF-β1 from HCPNs is likely due to electrostatic interactions between negatively charged sulfate groups in heparin and the positively charged amino acid residues in TGF-β1 protein (20). Growth factor protein loading in polymeric delivery systems often results in loss of the growth factor bioactivity. This is due to denaturation of growth factor protein exposed to harsh environments, such as heating, sonication, and organic solvents, during the process of protein loading in polymer carrier (2). Fortunately, our system released bioactive TGF-β1, as TGF-β1 released from HCPNs induced expression of cardiomyogenic markers in BMSCs, which was similar to that induced by TGF-β1 daily added to the medium of BMSC culture (Fig. 3). This was likely due to TGF-β1 loading to HCPNs without exposure to harsh environments such as sonication and organic solvents.
Local delivery of TGF-β1 induced in situ cardiomyogenic differentiation of BMMNCs implanted into ischemic myocardium. Previous studies have shown that TGF-β1 induce cardiomyogenic differentiation of CD117+ bone marrow stem cells in vitro (10,11). TGF-β1 induced mRNA expression of cardiac MHC, troponin-I, troponin-T, and connexin 43 in CD117+ bone marrow stem cells in vitro after 3 days of cultivation (10). Immunocytochemical analysis showed that 60%, 24%, and 13% of TGF-β1-stimulated CD117+ bone marrow stem cells were cardiac MHC, troponin-I, and connexin 43 positive after 28 days of culture (11). However, CD117+ bone marrow stem cells that were preprogrammed with TGF-β1 and implanted into infarcted myocardium of mice expressed only a myogenic marker (myosin) and did not express cardiomyogenic markers (10). Our method was successful in inducing expression of cardiomyogenic markers (i.e., cardiac MHC, connexin 43, and α-sarcomeric actin) in BMMNCs implanted into infarcted myocardium (Fig. 5–7). This could be due to TGF-β1 that was delivered locally to the cell implantation site over a long period. The in situ differentiation method developed in this study would be advantageous for clinical applications over the in vitro culture method for cardiomyogenic differentiation, since urgent patients with myocardial infarction do not need to wait during the culture period and can be treated promptly.
TGF-β1 can induce osteogenic differentiation of BMMSCs depending on the concentration of TGF-β1 (10). A study has shown that TGF-β1 at a low concentration (5 ng/ml or less) did not induce osteogenic differentiation of CD117+ bone marrow cells in vivo or in vitro, but TGF-β1 at a higher concentration (100 ng/ml) did (10). TGF-β1 delivery at an inappropriate concentration may induce bone formation within the myocardium. Fortunately, TGF-β1 delivery using our system did not induce osteogenic differentiation of BMMNCs (Fig. 8). In our system, 2 ng of TGF-β1 loaded on HCPNs in 0.1 ml of fibrin was injected into myocardium, and the TGF-β1 was released into myocardium presumably for several weeks. No osteogenic differentiation of BMMNCs in myocardium (Fig. 8) was likely due to the maintenance of TGF-β1 at a low concentration at the myocardium.
Conclusion
HCPNs suspended in fibrin gel elicited a sustained release of bioactive TGF-β1 in vitro. In situ cardiomyogenic differentiation of BMMNCs implanted in an undifferentiated state to infarcted myocardium was induced by local delivery of TGF-β1 to the cell implantation site, and improved cardiac functions. This technology would significantly improve stem cell therapy for myocardial infarction, as it can remove in vitro stem cell culture step for cardiomyogenic differentiation which is costly and laborious and requires patients to wait for several weeks.
Footnotes
Acknowledgments
This work was supported by the Korea Health 21 R&D Project, Ministry of Health, Welfare, and Family Affairs (grant number: A050082), the Stem Cell Research Center of the 21th Century Frontier Program (grant number: SC3220), the National Research Foundation of Korea (grant number: 2010-0020352), and Seoul Science Fellowship (Hee Seok Yang), Republic of Korea. The authors declare no conflicts of interest.