
Abstract
Increasing amounts of human hepatocytes are needed for clinical applications and different fields of research, such as cell transplantation, bioartificial liver support, and pharmacological testing. This demand calls for adequate storage options for isolated human liver cells. As cryopreservation results in severe cryoinjury, short-term storage is currently performed at 2–8°C in preservation solutions developed for the storage of solid organs. However, besides slowing down cell metabolism, cold also induces cell injury, which is, in many cell types, iron dependent and not counteracted by current storage solutions. In this study, we aimed to characterize storage injury to human hepatocytes and develop a customized solution for cold storage of these cells. Human hepatocytes were isolated from material obtained from partial liver resections, seeded in monolayer cultures, and, after a preculture period, stored in the cold in classical and new solutions followed by rewarming in cell culture medium. Human hepatocytes displayed cold-induced injury, resulting in >80% cell death (LDH release) after 1 week of cold storage in University of Wisconsin solution or cell culture medium and 3 h of rewarming. Cold-induced injury could be significantly reduced by the addition of the iron chelators deferoxamine and LK 614. Experiments with modified solutions based on the new organ preservation solution Custodiol-N showed that ion-rich variants were better than ion-poor variants, chloride-rich solutions better than chloride-poor solutions, potassium as main cation superior to sodium, and pH 7.0 superior to pH 7.4. LDH release after 2 weeks of cold storage in the thus optimized solution was below 20%, greatly improving cold storage of human hepatocytes. The results were confirmed by the assessment of hepatocellular mitochondrial membrane potential and functional parameters (resazurin reduction, glucagon-stimulated glucose liberation) and thus suggest the use of a customized hepatocyte storage solution for the cold storage of these cells.
Introduction
At present, a rising demand for human hepatocytes is experienced in clinical applications and several fields of research. Since there is a persistent shortage of donor organs, efforts are conducted towards alternative therapy options replacing liver transplantation, one of which is liver cell transplantation. The basic idea is to repopulate the diseased liver with functional cells infused either directly into the liver or into the spleen (34,56,59,69,74). In bioartificial liver support, extracorporally applied liver cells are used to support the liver functions that cannot be replaced by artificial detoxification alone (37–40,55,57). Finally, the use of human hepatocytes for drug testing and toxicity assays is increasing. All these applications require adequate storage strategies for isolated human liver cells, especially since the availability of human tissue is unpredictable and hepatocyte isolation is a complicated procedure that can only be performed in special centers with experienced staff (6,28). In the case of a successful cell isolation, large quantities of cells are available that are often too numerous for instant utilization.
Isolated hepatocytes are currently either cryopreserved or stored at 2–8°C (12). Although cryopreservation allows for virtually unlimited storage time, hepatocytes are very sensitive to cryoinjury, resulting in major cell loss (20,63,64) and severely reduced cell attachment (20,65). Cold storage at 2–8°C is usually performed under similar conditions as whole organ storage (1,24,60) in specially designed preservation solutions, such as University of Wisconsin (UW) solution. These solutions are adapted to the intracellular ion composition, possess a high buffering capacity, and contain impermeable substances (4,62). The time span for this kind of preservation for whole organs is limited to several hours; storage of isolated hepatocytes in these solutions is performed for a few days at maximum.
Although hypothermia has a protective effect by slowing down the cell metabolism, cold itself inflicts cell and organ damage (42,43,50,53,54), which is often amplified by subsequent rewarming (3,26,50). Cold-induced injury in several cell types has been shown to be caused by an increase in the intracellular chelatable iron pool followed by formation of reactive oxygen species (ROS) (42,43,45,48); this injury could be inhibited by addition of iron chelators to the preservation solution. In rat hepatocytes a second iron-independent but chloride-dependent pathway has been observed (44).
In this experimental study, we assessed whether these mechanisms of cold-induced injury also apply for cultured primary human hepatocytes and attempted to develop a customized preservation solution for this cell type.
Materials and Methods
Chemicals and Solutions
UW solution was purchased from Bristol-Myers Squibb (Viaspan; Princeton, NJ, USA), deferoxamine mesilate from Novartis (Basel, Switzerland), glucagon from Bachem Biochemica GmbH (Heidelberg, Germany), and MitoTracker Green from Molecular Probes (Eugene, OR, USA). LK 614 and N-acetylhistidine were kindly provided by Dr. F. Köhler Chemie (Bensheim, Germany). Williams' medium E and supplements were obtained from Biochrom KG (Berlin, Germany) and dexamethasone and glycine from Serva (Heidelberg, Germany). Modified preservation solutions (see Table 1 for composition) are based on the new organ preservation solution Custodiol-N (51,58) and some of them were already tested for the preservation of porcine aortic segments (70). The solutions were prepared in our laboratory and were supplemented as described in the respective tables and figure legends. Tetramethylrhodamine methyl ester (TMRM), adenosine, glucose oxidase, peroxidase, resazurin, and 2,2′-bipyridyl were obtained from Sigma-Aldrich (Taufkirchen, Germany). All other chemicals were acquired from Merck KGaA (Darmstadt, Germany).
Composition of Preservation Solutions

All concentrations are given in mmol/L unless stated otherwise. GSH, glutathione (reduced form); HES, hydroxyethyl starch; MOPS, 3-(N-morpholino)propanesulfonic acid.
Some of the solutions were already used in Wille et al. (70), but with different numbering (solution 6 in this article corresponds to solution 8 in Wille et al.).
Williams' medium E contains amino acids, trace elements, and antioxidants not listed in this table to maintain lucidity and was additionally supplemented with 10% FCS, streptomycin, HEPES, glutamine, and pyruvate (for concentrations refer to Materials and Methods) prior to use.
Calculated osmolarity.
Cell Isolation, Cell Culture, and Shipping
Human hepatocytes were isolated from tissue procured by partial liver resections performed at the clinic for general, visceral, and transplantation surgery, Charité-Universitätsmedizin Berlin. All donors underwent liver surgery for primary or secondary liver tumors. Written consent was obtained from all tissue donors and the protocol was approved by the local ethics committee. The specimen for cell isolation was taken from the tumor-free margin of the resected liver tissue.
Hepatocytes were isolated using a method of collagenase digestion as described previously (67). Briefly, the liver specimen was perfused via truncated larger vessels. In a first step, the tissue was flushed with a buffered solution containing 2.5 mmol/L ethylene glycol-bis(2-aminoethylether)-N,N,N′, N′-tetraacetic acid (EGTA) at 37°C to remove residual blood and destabilize the cell-to-cell connections. The liver tissue was digested for 17 ± 1 min (mean ± SEM) by recirculating perfusion (after a short flush) with the second buffered solution containing 1.4 U/ml collagenase P (Roche Diagnostics GmbH, Mannheim, Germany) and Ca2+. After mechanical disruption and filtering through a gauze-lined funnel, hepatocytes were washed in phosphate-buffered saline (PBS), centrifuged at 50 times; g and 4°C for 5 min, resuspended in PBS, and purified by density gradient centrifugation using a 25% Percoll solution (20 min at 1278 times; g; Percoll density: 1.124 g/ml, Biochrom AG, Berlin, Germany). Finally, cells were washed again in PBS, centrifuged at 50 times; g and 4°C for 5 min, and resuspended in culture medium (see below). Cells were then counted and viability was determined by trypan blue exclusion. Mean trypan blue exclusion before and after Percoll purification were 63 ± 3% and 76 ± 3%, respectively.
Cells were seeded onto collagen-coated six-well plates at 1 times; 106 living cells per well in Williams' medium E supplemented with 32 U/L insulin, 0.9 mg/L dexamethasone, 2 times; 105 U/L penicillin, 100 mg/L streptomycin, 1 mmol/L sodium pyruvate, 15 mmol/L N-2-hydroxyethylpiperazine-N′-2–ethane sulfonic acid (HEPES), 4 mmol/L L-glutamine, and 10% fetal calf serum (v/v) prior to use. After 10–16 h of culture, the culture medium was exchanged; the plates were sealed with sterile adhesive foil (SteriDrape, 3M, Neuss, Germany) and packed into a Styrofoam box containing gel pads heated to 37°C and a temperature logger (ThermaData logger, ETI Ltd., Worthing, UK) to trace the temperature changes during transport. Plates were then shipped to the lab overnight. At receiving the plates, the adhesive foil was removed under sterile conditions, and plates were checked for shipping damage or cell detachment. The culture medium was exchanged and cells were cultured for another 8–20 h before starting the experiment.
Cold Storage and Rewarming
At the start of the experiment, the cells were washed with Hanks' balanced salt solution (HBSS) at room temperature, the culture medium was changed to the respective cold storage medium (also at room temperature) supplemented or not with deferoxamine and/or LK 614 (see below). The plates were kept at 4°C for 168–504 h (1–3 weeks; cold storage time was increased with increasing protection to get clear-cut differences between solution variants). Culture plates with Williams' medium E (bicarbonate buffered) were kept in gas-tight containers that were flushed with 5% CO2/95% air.
After the respective cold storage time, a sample of the incubation solution was taken immediately after removing the plates from the cold to determine lactate dehydrogenase (LDH) release during cold storage. The wells were then thoroughly washed with cold HBSS, cold culture medium was added, and the plates were placed in an incubator at 37°C and 5% CO2. After 1, 2, and 3 h of rewarming, samples were taken from the culture medium. At the end of the experiment, the supernatant was removed and cells were lysed by addition of 1% (w/v) Triton X-100 in HBSS to determine residual intracellular LDH. Released LDH activity was given as percentage of total LDH activity (total LDH activity = released + intracellular LDH activity).
Assessment of Cell Death
LDH activity was measured photometrically with a standard assay of pyruvate-dependent NADH oxidation using a clinical chemistry analyzer (VITALAB Selectra E, Vital Scientific NV, Dieren, NL). LDH release at any given time point is expressed as percentage of total LDH activity of the respective well.
Resazurin Conversion
After 1 and 2 weeks of cold storage in UW solution or solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 and 30 min of rewarming in cell culture medium, cells were washed with HBSS, and 40 μM resazurin in warm HBSS + 10 mM D-glucose was added to the wells. Reductive conversion to resorufin was detected in a plate reader (FluoStar Optima, BMG Labtec, Offenburg, Germany) at λexc. = 560 + 10 nm and λem. ≥ 590 nm and 37°C for 10 min and conversion rate was calculated as percentage of warm control.
Glucose Liberation
After 1 and 2 weeks of cold storage in UW solution or solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 and 30 min of rewarming in cell culture medium, cells were washed with HBSS, and Krebs-Henseleit buffer with 20 mM lactate and 10 μg/L glucagon was added to the wells. After 4-h incubation at 37°C and 5% CO2, glucose concentration was determined in the supernatant by an enzymatic assay based on glucose oxidase adapted from Fossati et al. (13). Briefly, 100 μl of supernatant were added to 160 μl of reagent containing 5 mM 3,5-dichloro-2-hydroxy-benzenesulfonic acid, 4 mM 4-aminoantipyrine, 3 U glucose oxidase, 2 U peroxidase, and 100 mM KCl in 100 mM MOPS buffer (pH 6.7) and incubated at room temperature for 30 min. Formation of the pink product was determined in a plate reader at 550 nm. Glucose liberation represents the sum of gluconeogenesis and glycogenosis, with gluconeogenesis likely to be predominant in the presence of excess lactate.
Confocal Laser Scanning Microscopy
Cells were cultured on collagen-coated glass cover slips in six-well plates and loaded for 40 min with 500 nM MitoTracker Green and 20 min with 500 nM TMRM at 37°C (warm control). Cells in the cold were incubated with MitoTracker during the complete cold incubation time (336 h); prolonged incubation times with the dye did not have any adverse effects on cell viability. The cold storage solution was exchanged 60 min before the end of cold incubation for 500 nM TMRM in cold HBSS. After loading, the medium was changed to HBSS with 200 nM TMRM (maintenance dose). Fluorescence was assessed by confocal laser scanning microscopy using a Zeiss Axiovert 100M microscope with a LSM 510 laser scanning module (Zeiss, Oberkochen, Germany). For TMRM and MitoTracker Green detection, λexc = 543 nm (helium/neon laser), λem ≥ 585 nm and λexc. = 488 nm (argon laser), λem. = 505–530 nm were used, respectively.
Statistics
All experiments were performed in duplicates and repeated 3–12 times (see numbers given in the respective tables/figure legends). In box plots and tables, data are expressed as median and 25/75 percentiles (whiskers represent 1.5 interquartile range and outliers are marked as dots). Box plots were created using R (Version 2.6.2, Copyright: The R Foundation for Statistical Computing), bar graphs were generated in GraphPad Prism (GraphPad Software Inc., La Jolla, CA, US). Since the variation between different donors was expected to be high, only pairs/sets from identical donors were used for comparison (hence the differing numbers for different experiments). Due to the small sample size, the nonparametric Wilcoxon matched-pairs signed-rank test was performed to assess differences between two groups after cold storage and subsequent rewarming. For comparing two groups of data on functional parameters, paired t-test was employed. A value of p ≤ 0.05 was considered as statistically significant.
Results
Cold-Induced Cell Injury to Human Hepatocytes
Primary human hepatocytes showed cold-induced injury when stored at 4°C, which was dependent on cold storage time (Fig. 1 and data not shown). Part of the cold-induced injury only became evident during rewarming. Cold-induced damage could be observed in cell culture medium (Fig. 1A, B) but also in the organ preservation solution UW (Fig. 1C, D).

Protective effect of the iron chelator deferoxamine against cold-induced injury of human hepatocytes. Human hepatocytes were stored in the cold (4°C) in culture medium (A, B) or University of Wisconsin solution (UW; C, D) with or without deferoxamine, respectively, for 1 week (A, C) and 2 weeks (B, D) and rewarmed for 3 h (37°C) in cell culture medium. (A) p = 0.016 for 171 h, n = 7; (B) p = 0.063 for 339 h, n = 6; (C) p = 0.015 for 171 h, n = 7, (D) p = 0.031 for 339 h, n = 6.
Effect of Iron Chelators
To assess whether or not cold-induced cell injury in human hepatocytes is iron dependent, iron chelators were added. The iron chelator deferoxamine decreased cold-induced injury after 1 week of cold storage in cell culture medium and 3 h of rewarming from 90% (median for cell culture medium after 171 h) to 26% (median for cell culture medium + 1 mM deferoxamine after 171 h) (Fig. 1A). A similar protective effect could be observed with the iron chelator 2,2′-bipyridyl (data not shown), evidencing that cold-induced cell injury is iron dependent in human hepatocytes. Addition of iron chelators also decreased cell damage in UW solution after 1 week of cold storage and 3 h of rewarming (Fig. 1C). After 2 weeks of cold storage, partial protection could still be observed in UW solution (Fig. 1D), but not in cell culture medium (Fig. 1B).
Improvement of the Base Solution
The above shown influence of the base solution (cell culture medium vs. UW solution, in the presence and absence of iron chelators) on cell protection in the cold suggested that modification of the base solution might further enhance protection. Therefore, we further varied the composition of the base solution, resorting to previous experience with cold storage of different cell types.
Comparing base solutions with different ion compositions, the ion-rich variants (solution 1 and 2, Table 1) showed better protection compared to the ion-poor variant (solution 3, Tables 1 and 2). Replacing MOPS (in solution 3) by N-acetylhistidine (solution 4) (70) did not affect cell injury (data not shown).
Influence of the Ion Compositions of the Storage Solution on Cold-Induced Injury of Human Hepatocytes

Human hepatocytes were stored in the cold for 2 weeks and subsequently rewarmed for 3 h (n = 8). The table shows LDH release as percentage of total LDH (group median and 25%/75% confidence intervals). The nonparametric Friedmann test with Dunn post hoc test showed a significant difference between solution 2 and solution 3 (p < 0.01).
Based on these findings, solution 5 (Table 1) is an ion-rich variant containing N-acetylhistidine as buffer. With regard to the anion, solution 5 can be seen as a successor solution to solution 2, but it contains potassium as main cation because the advantage of potassium over sodium was observed in rat hepatocytes (verified later for human hepatocytes, see below). Two weeks of cold incubation in solution 5 with 1 mM deferoxamine followed by 3 h of rewarming resulted in only 20% LDH release (Fig. 2A), representing a major improvement compared to cell culture medium and UW solution. Even after 3 weeks of cold storage in solution 5 with 1 mM deferoxamine and subsequent rewarming, LDH release was still below 50% (Fig. 2B).
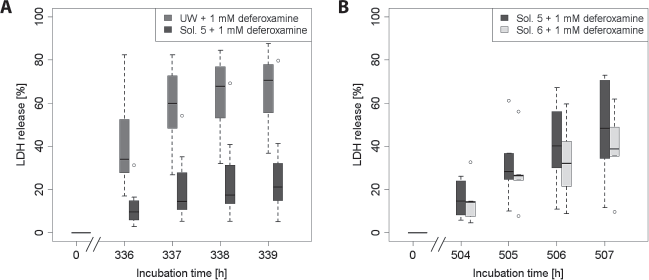
Improved hepatocyte preservation with the new storage solutions. (A) Comparison of solution 5 + 1 mM deferoxamine with UW + 1 mM deferoxamine (solution 5 is Cl- rich, contains potassium as main cation, and has a pH of 7.0) (Table 1). Cells were stored at 4°C for 2 weeks (336 h), followed by 3 h of rewarming. After 339 h, the difference in median between the two groups is statistically significant (n = 7; p = 0.016). (B) Comparison of solution 5 + 1 mM deferoxamine and solution 6, the latest version of the optimized preservation solution, + 1 mM deferoxamine (n = 6; p = 0.063). Cells were stored in the cold for 3 weeks (504 h) and subsequently rewarmed for 3 h.
The amino acid composition was then adapted to the new organ preservation solution Custodiol-N (51,58) developed in our group. The resulting solution, solution 6 (Table 1, minor changes compared to solution 5), offered slightly better protection than solution 5 for 3 weeks of cold incubation and 3 h of rewarming (Fig. 2B).
Role of Chloride
Previous experiments in rat hepatocytes suggested that the extracellular chloride concentration plays a prominent role in cold-induced cell damage (44). To assess the role of chloride in cold-induced injury of human hepatocytes, chloride-poor variants of solution 5 (solution 7) and solution 6 (solution 8) were used, in which chloride was almost completely replaced by the impermeant anion lactobionate (Table 1). As solution 6 had shown excellent results in the previous series (Fig. 2B), cold incubation time was prolonged to 3 weeks for the comparison of solution 6 and solution 8 in order to amplify the potential differences between these solutions. Cell damage during cold storage itself was identical for Cl--poor and Cl--rich solutions in both pairs of solutions, but cell damage was increased during rewarming after cold storage in Cl--poor solutions when compared to cold storage in Cl--rich solutions [Fig. 3A (p = 0.219 for 339 h) and Fig. 3B (p = 0.016 for 507 h)].

Influence of chloride on cold-induced injury of human hepatocytes. Cold storage of human hepatocytes (4°C) in chloride-rich solutions (solutions 5 and 6) or corresponding solutions in which lactobionate was used as a substitute for Cl- (solutions 7 and 8) for 2 weeks (336 h) or 3 weeks (504 h) followed by 3 h of rewarming (37°C). The difference in lactate dehydrogenase (LDH) release for (B) is statistically significant after 3 h of rewarming (n = 7, p = 0.016).
Optimization of the Base Solution
To verify the effects of single components in the final storage solution, solution 6, the impacts of further clear-cut modifications were assessed (again after 3 weeks of cold storage). When comparing solution 6 (potassium as main cation) and solution 9 (sodium as main cation, Table 1), survival after cold storage in the potassium-rich solution 6 was significantly better than after cold storage in the sodium-rich solution 9 (Table 3). Furthermore, cell death after 3 weeks of cold storage and 3 h of rewarming was significantly reduced in solution 6 (pH 7.0) compared to solution 10 (pH 7.4, Table 3). Supplementation of solution 6 + 1 mM deferoxamine with 5 mM adenosine appeared to lead to some further improvement, but the effect was not quite significant (Table 3). Next, the combination of hydrophobic and hydrophilic iron chelators was assessed. With 20 μM of the hydrophobic iron chelator LK 614 and 0.5 mM of deferoxamine, the same protective effect as seen with 1 mM deferoxamine could be achieved (Table 3).
Influence of Further Features of the Cold Storage Solution on Cold-Induced Injury to Human Hepatocytes

Cells were stored in the cold for 3 weeks and subsequently rewarmed for 3 h. The table shows LDH release as percentage of total LDH (group median and 25%/75% confidence intervals). Wilcoxon matched-pair signed-ranks test was performed to compare each two associated groups.
Thus, the least cold-induced cell damage in human hepatocytes was observed in chloride- and potassium-rich solution 6 at pH 7.0 with 1 mM deferoxamine or 0.5 mM deferoxamine and 20 μM LK 614 (with or without adenosine). Solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 was then used for the assessment of further parameters of cell integrity.
Cell Morphology After Cold Storage and Rewarming
Before the experiments, hepatocyte cultures showed normal monolayer morphology (Fig. 4A). After 2 weeks of cold storage in UW, cells displayed severe granulation of the cytoplasm, occasional detachment, vacuolization, and nuclear condensation directly after the cold incubation (Fig. 4B), and additional blebbing during rewarming (Fig. 4C). After 2 weeks of cold storage in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614, cells had retracted on the cell culture surface but did not detach (Fig. 4D). They did not display increased granulation of the cytoplasm nor blebbing at rewarming. Cell retraction was already partially reversed after 0.5 h of rewarming in cell culture medium (Fig. 4E); after 3 h of rewarming respreading was complete (data not shown), thus underlining that cellular viability was preserved.

Cell morphology after 2 weeks of cold storage and rewarming. Cells were stored at 4°C in University of Wisconsin (UW) solution (B, C) or in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 (D, E) for 2 weeks and rewarmed in cell culture medium for 30 min at 37°C and 5% CO2. Phase contrast microscopy was performed before cold storage (warm control, A), directly after cold storage (B, D) and after rewarming (C, E). Note the granulation of cells in (B) and (C) (long arrows), the condensation of nuclei (arrowheads) and bleb formation in (C) (short arrows).
Preservation of Mitochondrial Integrity and Function
Mitochondrial alterations during cold storage and rewarming were assessed using two fluorescent dyes, TMRM, which accumulates in mitochondria depending on membrane potential, thus only staining intact mitochondria (red fluorescence), and MitoTracker Green, which binds covalently and stays in the mitochondria even after loss of membrane potential (green fluorescence). Control cells displayed numerous mitochondria filling almost the whole cell (Fig. 5A). After 48 h of cold storage, mitochondrial membrane potential was largely preserved, irrespective of the storage solution (Fig. 5B–D), but after 2 weeks of cold storage, a total loss of mitochondrial membrane potential (control: Fig. 6A) was seen in cells stored in UW (Fig. 6B). However, most of the cells maintained their mitochondrial membrane potential when stored in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 (Fig. 6C). Cells stored in solution 8 with 0.5 mM deferoxamine and 20 μM LK 614 for 2 weeks partially retained mitochondrial membrane potential after 3 h of rewarming (Fig. 6D), but the proportion of cells in which most mitochondria lost their membrane potential was higher than that of cells stored in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614.

Mitochondrial membrane potential and morphology of human hepatocytes after 48 h of cold storage and rewarming. Cells were labeled with 500 nM tetramethylrhodamine methyl ester (TMRM) for 20 min (37°C, control) and 60 min (4°C) and visualized using confocal laser scanning microscopy. (A) Warm control (37°C). (B, C) Cells after 48 h of cold storage (4°C) in solution 6 + 0.5 mM deferoxamine + 20 μM LK 614 (B) and 3 h of rewarming in culture medium (C). (D) Cells after 48 h of cold storage in UW and 3 h of rewarming in cell culture medium.

Mitochondrial membrane potential and morphology of human hepatocytes after 2 weeks of cold storage and rewarming. Cells were labeled with 500 nM MitoTracker Green for 40 min (control, 37°C) or 2 weeks (in the cold, 4°C) and 500 nM TMRM for 20 min (control) or 60 min (4°C) and visualized using confocal laser scanning microscopy. Control (37°C, A); cells after 2 weeks of cold incubation in UW (B), solution 6 + 0.5 mM deferoxamine + 20 μM LK 614 (C) and solution 8 + 0.5 mM deferoxamine + 20 μM LK 614 (D) and 3 h of rewarming in cell culture medium.
Assessment of mitochondrial morphology showed filamentary branched mitochondria in control cells (Fig. 5A), but a fragmentation of mitochondria directly after cold storage in all solutions (Fig. 5B for solution 6 with 0.5 mM deferoxamine and 20 μM LK 614). Mitochondrial fragmentation was reversible after 3 h of rewarming in cell culture medium when cells were stored in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 for 48 h (Fig. 5C), while the morphological changes were irreversible in cells stored in UW (Fig. 5D). Mitochondrial fragmentation after the extensive cold storage period of 2 weeks was still partially reversible in cells stored in solutions 6 and 8 (Fig. 6C, D, inset), but noticeable swelling of mitochondria was seen in cells stored in UW (Fig. 6B).
Cellular Reductive Capacity
After 1 week of cold storage in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614, the conversion of resazurin to resorufin as marker for reductive cell metabolism (33) was not decreased compared to control cells (Fig. 7), while reductive metabolism of cells stored in UW solution only amounted to about 50% of initial value. After 2 weeks of cold storage, reductive capacity of cells stored in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 was slightly decreased (compared to unstored warm controls) while it was below 10% for cells stored in UW.

Reductive capacity of human hepatoyctes after 1 and 2 weeks of cold storage and rewarming. Cells were stored at 4°C for 1 week (n = 3) and 2 weeks (n = 4) and rewarmed in cell culture medium for 30 min at 37°C and 5% CO2. Thereafter, the reduction of resazurin to resorufin was tested fluorimetrically. All values are expressed as percentage of the respective warm control (mean ± SD). Asterisks indicate significant difference between the two groups as determined with the paired t-test.
Glucose Liberation
Glucagon-induced glucose liberation during incubation with 20 mM lactate, as marker of gluconeogenesis, only slightly decreased during 1 week of cold storage in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 to about 90% of the value of warm, unstored control cells (Fig. 8). Even after 2 weeks of cold storage in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614, glucagon-induced glucose liberation remained at about 60% of controls, while it was already below 25% after 1 week of cold storage in UW solution and below 10% after 2 weeks of cold storage in UW solution, showing that hepatocyte-specific metabolic function of cells stored in solution 6 + 0.5 mM deferoxamine and 20 μM LK 614 was significantly better than that of cells stored in UW solution after 2 weeks of cold storage.

Glucagon-stimulated glucose liberation of human hepatoyctes after 1 and 2 weeks of cold storage and rewarming. Cells were stored at 4°C for 1 week (n = 3) and 2 weeks (n = 4) and rewarmed in cell culture medium for 30 min at 37°C and 5% CO2. Thereafter, cells were washed with HBSS and incubated with KH + 20 mM lactate and 10 μg/L glucagon for 4 h to assess gluconeogenesis. Glucose concentration was determined in the supernatant after 4 h. All values are expressed as percentage of the respective warm control (mean ± SD). Asterisks indicate significant difference between the two groups as determined with the paired t-test.
Discussion
We here showed that human hepatocytes suffer iron-dependent cold-induced injury when stored at 4°C and that the rate of cell survival (Figs. 2 and 3) as well as cell morphology (Fig. 4) and metabolic function (Figs. 7 and 8) could be largely enhanced by using an optimized storage solution.
Iron-Dependent Cold-Induced Injury in Human Hepatocytes
Human hepatocytes display iron-dependent (i.e. iron chelator-inhibitable) cold-induced injury after cold incubation at 4°C and during subsequent rewarming (Fig. 1). In earlier publications with other cell types, we showed that cold-induced cell injury is triggered by “chelatable” or “redox-active” cellular iron ions (26,48). During cold storage, a massive increase in cytosolic or cellular chelatable iron has been observed (22,26,48), triggering oxidative injury including lipid peroxidation via formation of highly reactive oxygen species and subsequent mitochondrial injury (25,43,45,49,54). Our results show that human hepatocytes suffer injury of a similar mechanism, where the mitochondria also appear to be a major target (Figs. 5 and 6).
Classical Mechanisms of Cold-Induced Injury
The classical view of the mechanism of cold-induced injury is that cold-induced inhibition of Na+/K+-ATPase leads to a pronounced cellular sodium accumulation accompanied by Cl- accumulation and water uptake, which leads to cell swelling and finally cell death (4,21). Based on this theory, most organ preservation solutions contain low sodium and chloride concentrations (4,5). However, Fuckert et al. (16) and Gizewski et al. (17) showed that both cytosolic sodium concentration and intracellular sodium and chloride content decreased during normoxic hypothermia in liver endothelial cells and hepatocytes. In the latter cells, sodium accumulation was only triggered by hypoxia and not by hypothermia. Furthermore, cold-induced injury in these cells is of an apoptotic rather than necrotic type (50,54) and thus associated with cell shrinkage and not with cell swelling (52) as proposed in the classical mechanism.
The Role of Chloride in Cold-Induced Injury
Although the classical mechanism does not appear to be relevant in rat hepatocytes, these cells still display a certain amount of chloride-dependent, but sodium-independent cold-induced injury, which is also iron independent (44). This injury is reduced in chloride-free solutions. However, in human hepatocytes a chloride-free solution did not decrease but rather increase cold-induced cell injury (Fig. 3B). Thus, the behavior of human hepatocytes is contrary to that of rat hepatocytes, but similar to porcine aortic endothelial cells (70), which also show decreased damage in chloride-rich storage solutions.
Cell Morphology
Cells stored in solution 6 with 0.5 mM deferoxamine and 20 μM LK 614 showed marked retraction during cold storage, which was already seen in other cell types (46) and is reversible during rewarming. Retraction generally seems more marked in cells surviving cold incubation [(46) and U. Rauen, T. Noll, S. Knoop, unpublished results].
Mitochondrial Fragmentation
Imbalance of mitochondrial fission/fusion is a phenomenon related to cell stress in various cell types (9,29,31,61). Mitochondrial fission has previously been observed with cold storage of rat liver endothelial cells in UW solution (25). In these cells, shortening also occurred in the presence of an iron chelator, but was reversible on rewarming. Unlike the mitochondrial permeability transition and unlike mitochondrial ultracondensation, mitochondrial fission did not appear to be involved in the injurious process but proved to be reversible during rewarming (25,46) as observed here.
Components and Principles of the New Solution as Adopted From Custodiol-N
For cell storage, in most studies, organ preservation solutions are used (2,10). Since in the current study the composition of the base solution seems to have a major effect on the protection against cold storage injury (Fig. 1, Table 2), an optimized solution for human hepatocytes was developed. Starting point was the newly designed preservation solution Custodiol-N (51), which is based on the classical histidine-tryptophan-ketoglutarate (HTK) organ preservation solution and has already been tested for organ preservation in animal experiments (72).
In Custodiol-N, several components were altered and some additions made compared to HTK: the iron chelators deferoxamine and LK 614 were added to prevent iron-dependent cold-induced injury (51,73). Glycine and alanine inhibit the formation of a hypoxia-induced plasma membrane pore and prevent hypoxic injury (7,11,14); therefore, both small amino acids are present in Custodiol-N. Histidine was partially replaced by N-acetylhistidine, since we showed earlier that histidine, the major buffering compound in HTK solution, has a toxic potential in rat hepatocytes that is not shared by N-acetylhistidine (41,47). Sucrose replaces mannitol, as it is a superior impermeant (8,23,68). Moderate acidosis has been shown to provide protection (18,19); therefore, Custodiol-N displays a pH of 7.0. α-Ketoglutarate was inherited from the original HTK composition.
Optimization of the Hepatocyte Storage Solution
For cell storage, several alterations were made to Custodiol-N: histidine was completely replaced by N-acetylhistidine and the overall buffer concentration was reduced. The Ca2+ concentration was slightly raised in the new solution, because very low concentrations of calcium are harmful to hepatocytes (41,66). Glucose was added as additional energy substrate as previously described by Wille et al. (70).
Other components were evaluated separately in the development of the final hepatocyte storage solution: the advantage of pH 7.0 compared to higher pH values of the storage solution, which was suggested before in porcine aortic segments (70) also applies to primary human hepatocytes (Table 3). Potassium as main cation of preservation solutions is discussed controversially. Potassium is supposed to be cytotoxic, especially in endothelial cells (27,30,35), although, on the other hand, it has also been suggested to contribute to the protective effect of UW solution (4). Furthermore, in cell transplantation, potassium might cause cardiac arrhythmias when infused in higher amounts together with the cells. Still, for human hepatocytes as well as for porcine endothelial cells (70), cold-induced cell injury is decreased in potassium-rich solutions, most likely by the inhibition of cellular potassium efflux. In addition, as already discussed above, human hepatocytes benefit from high (physiological) chloride concentrations in the storage solution.
Adenosine is supposed to trigger receptor-mediated cytoprotective pathways in various cell types, although this was mainly shown under hypoxic conditions (15,32,36). By addition of adenosine, a slightly improved cell survival was achieved in human hepatocytes, but the difference was not quite significant. However, adenosine should be considered as potential additive and evaluated further.
Iron chelator concentrations were increased in the new solution compared to Custodiol-N, since cultured cells are more sensitive to cold-induced iron-dependent injury than tissue or whole organs [(70) and unpublished results]; as in other tissues the combination of deferoxamine and LK 614, preferable on theoretical grounds (quicker intracellular chelation by the more membrane-permeable but weaker LK 614), showed similar protection as higher concentrations of deferoxamine alone.
Conclusion and Perspective
Iron-dependent cold-induced cell injury does occur in human hepatocytes. The developed preservation solution (solution 6 + 0.5 mM deferoxamine + 20 μM LK 614), with potassium as main cation, chloride as main anion, the iron chelators deferoxamine and LK 614, and pH 7.0, allows prolonged cold storage of human hepatocytes, thus enhancing the time frame for cell transplantation or extracorporeal liver support by permitting cell shipping at 4°C without major loss of function. As the solution is based on the organ preservation solution/cardioplegic solution Custodiol-N (72), which has just entered clinical studies, and is similar to the vascular preservation solution TiProtec® (70,71), which is already approved in Europe for vascular storage (71), usage of solution 6 in the field of cell transplantation appears feasible. The solution will now be further evaluated in the specific settings for these clinical applications, especially focusing on storage of hepatocyte suspensions.
Footnotes
Acknowledgments
We thank Ms. N. Boschenkov and Mr. C. Fehring for their excellent technical support. This study was supported by the German Federal Ministry of Education and Research (BMBF–0312111/12). U. Rauen obtained consulting fees from Dr. Franz Köhler Chemie GmbH, Bensheim, Germany. Dr. F. Köhler Chemie GmbH holds a patent on the preservation solution used in this study.