
Abstract
When using human induced pluripotent stem cells (hiPSCs) to achieve hair follicle (HF) replacement, we found it best to emulate the earliest fundamental developmental processes of gastrulation, ectodermal lineage commitment, and dermogenesis. Viewing hiPSCs as a model of the epiblast, we exploited insights from mapping the dynamic up- and down-regulation of the developmental molecules that determine HF lineage in order to ascertain the precise differentiation stage and molecular requirements for grafting HF-generating progenitors. To yield an integrin-dependent lineage like the HF in vivo, we show that hiPSC derivatives should co-express, just prior to transplantation, the following combination of markers: integrins α6 and β1 and the glycoprotein CD200 on their surface; and, intracellularly, the epithelial marker keratin 18 and the hair follicle bulge stem cell (HFBSC)-defining molecules transcription factor P63 and the keratins 15 and 19. If the degree of trichogenic responsiveness indicated by the presence of these molecules is not achieved (they peak on Days 11-18 of the protocol), HF generation is not possible. Conversely, if differentiation of the cells is allowed to proceed beyond the transient intermediate progenitor state represented by the HFBSC, and instead cascades to their becoming keratin 14+ keratin 5+ CD200– keratinocytes (Day 25), HF generation is equally impossible. We make the developmental case for transplanting at Day 16-18 of differentiation—the point at which the hiPSCs have lost pluripotency, have attained optimal expression of HFBSC markers, have not yet experienced downregulation of key integrins and surface glycoproteins, have not yet started expressing keratinocyte-associated molecules, and have sufficient proliferative capacity to allow a well-populated graft. This panel of markers may be used for isolating (by cytometry) HF-generating derivatives away from cell types unsuited for this therapy as well as for identifying trichogenic drugs.
Keywords
Introduction
Regenerative Medicine seeks to use stem cells to replace cells that have undergone destruction or senescence and death. Among the most sought cell types are hair follicles (HFs) for patients with inherited or immunogenic alopecia, or alopecia following severe wounding (e.g., burns, trauma, surgery), or androgenetic alopecia 1 . There has long been a debate over what qualities a stem cell and its derivatives should possess to enable HF replacement 2,3 . We present here the developmental and molecular requirements that human pluripotent stem cells (hPSCs) and their derivatives must acquire to reconstitute HFs—whether using human embryonic stem cells (hESCs) or, more immunologically desirable, patient recipient-specific human induced pluripotent stem cells (hiPSCs). These insights into successful engraftment were gained by examining the dynamics of up- and down-regulation of the key molecules that ensure proper HF lineage commitment by hPSCs. We show that hPSCs intended for HF replacement should co-express the following combination of markers: key components of the integrin signaling pathway -- integrin α6 (ITGA6) 4,5 and integrin β1 (ITGB1) 6 –9 and, just prior to transplantation, the surface glycoprotein CD200 10,11 together with keratin 18 (KRT18) (an epithelial marker) 12 and the following hair follicle bulge stem cell (HFBSC) markers: transcription factor P63 5,13 , keratin 15 (KRT15) 5,14 , and keratin 19 (KRT19) 5,15,16 . CD200, ITGA6, and ITGB1 on the surface of the hPSC likely interact with specific matrix receptors that not only mediate cell adhesion, survival, and proliferation 17 , but also entrance into a differentiation pathway that allows the emergence of HFBSCs in vitro and HFs in vivo. If the developmental stage and degree of responsiveness indicated by the presence of these markers is not achieved, HF replacement will not be possible. In other words, to yield optimal HFs in vivo, we make the developmental case for transplanting at Day 16 of differentiation—the point at which the hPSCs have lost almost all expression of pluripotency markers; have attained optimal expression levels of HFBSC markers but not yet progressed toward a keratinocyte fate; have not yet experienced downregulation of key integrins and surface CD200; and possess sufficient proliferative capacity to produce a well-populated graft. In fact, the necessity of these markers may be used to separate hiPSC derivatives that will yield HFs away from a heterogeneous population of derivatives that are disadvantageous for this therapeutic indication or perhaps even inimical (i.e., tumorigenic). The markers may also be used to identify trichogenic drugs.
Material & Methods
Human Pluripotent Stem Cells (hPSCs)
Two types of hPSCs were used. For hESCs, the “gold standard” for hPSCs, we employed the H9 line (Wicell). We generated our own hiPSCs from normal human skin fibroblasts (obtained from de-identified donors) by using a Neon transfection system to introduce, by electroporation, non-integrating episomal plasmid vectors encoding OCT3/4, SOX2, KLF4, L-MYC, LIN28 and an shRNA for human p53 18 (see Supplemental Material).
Characterization of hiPSCs
As previously described 19 –21 , hiPSCs colonies were characterized in vitro by both immunocytochemistry (ICC) and flow cytometry (FC) for the presence of the following standard pluripotency markers using monoclonal antibodies against OCT4, NANOG, SOX2, TRA-1-60, TRA-1-81 & SSEA4. Pluripotency of the hiPSCs was further confirmed by proving their ability to form teratomas when injected into the flanks of immuno-incompetent (NSG) mice (Jackson Laboratory). The teratomas were examined histologically for the presence of cells representative of the 3 fundamental primitive germ layers (ectoderm, mesoderm, and endoderm), hence confirming pluripotency (see Supplemental Material).
Differentiation of hiPSCs into HFBSCs
As a continuation of our previous work 22 , in this article, our overall strategy was to differentiate hiPSCs toward becoming keratinocytes but, just prior to that end-point, capture and isolate an intermediate transient progenitor cell population in vitro that expressed “HFBSCs” markers and could generated HFs. As schematized in Fig. 1 and detailed under Results and in the Supplemental Material, the protocol was divided into 2 parts: first, the formation of embryoid bodies (EBs) in a floating culture followed, second, by plating the EBs onto collagen I coated plates. The protocol was repeated with at least 10 technical replications on at least 2 different hiPSC clones. To differentiate hiPSCs into keratinocytes, we used a sequential differentiation protocol that employs all-trans retinoic acid (ATRA) and L-ascorbic acid (L-AA) to induce hiPSC to form ectoderm, which were then differentiated into HFBSCs via the addition of bone morphogenic protein-4 (BMP-4) and epidermal growth factor (EGF). In this protocol, ectoderm is precluded from continuing to become neuroectoderm via BMP-4 suppression (see Supplemental Material).
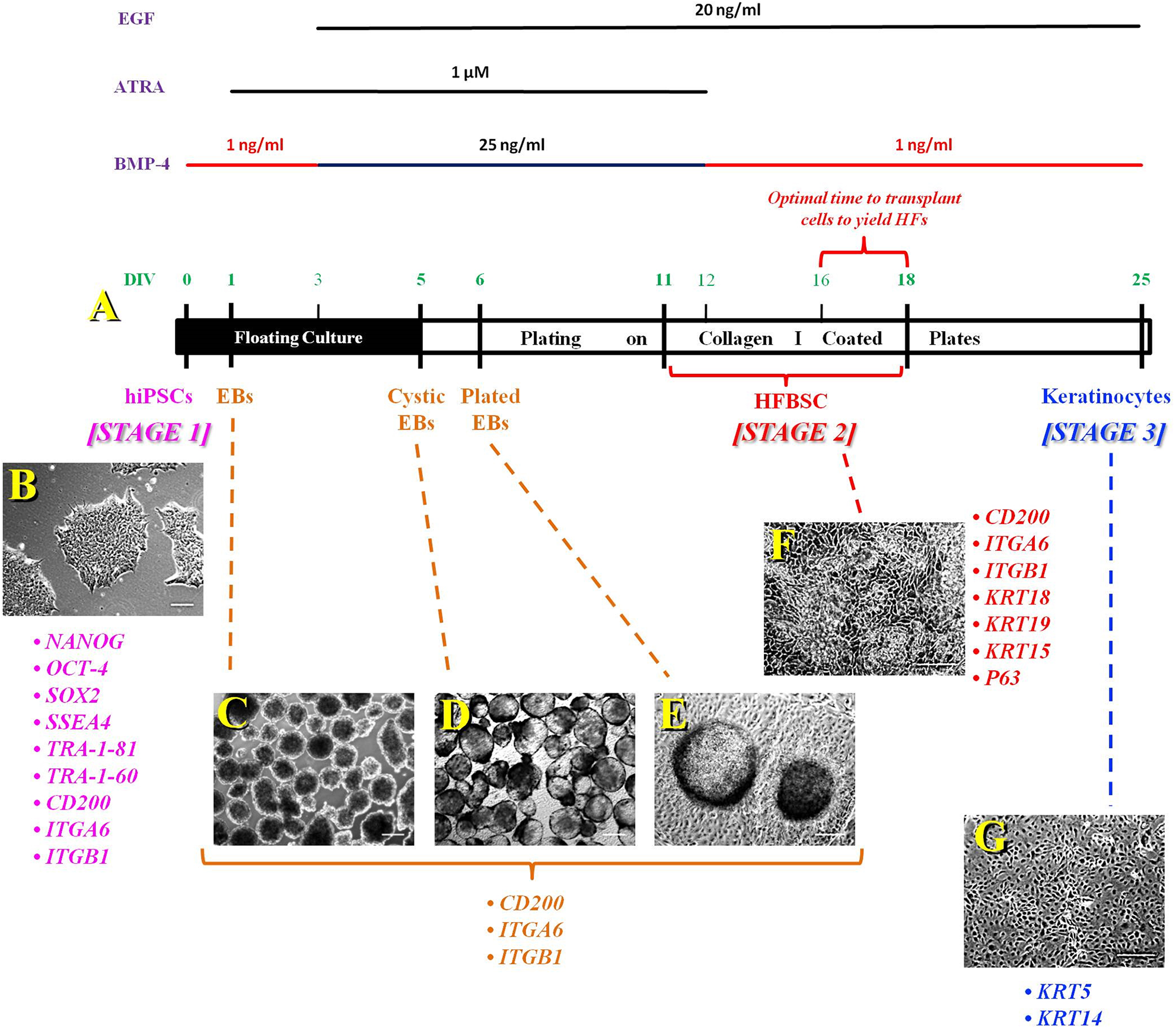
Protocol developed to generate HFBSCs and keratinocytes from hiPSCs. (A) The protocol is divided into 2 parts: First, formation of floating EBs; second, plating of the EBs on collagen type I-coated plates. Throughout the differentiation protocol, the differentiated cells have 3-stage profile: Stage #1 is starting with the undifferentiated hPSCs, either hESCs or, as illustrated here, hiPSCs. Stage #2, the intermediate transient progenitor stage comprised of HFBSCs. These cells are the ones to be harvested and transplanted to yield HFs in vivo. If they are not harvested, then they will continue to differentiate into keratinocytes (Stage #3) which have lost the competence to yield HF. To differentiate hiPSCs into keratinocytes, we used this sequential differentiation protocol that employs ATRA and L-AA to induce hiPSC to form ectoderm, which were then differentiated into HFBSCs via the addition of BMP-4 and EGF. In this schematic, the time line for the appearance of each stage is shown, along with representative photomicrographs of cells at each stage (B-G), illustrating the morphological changes undergone by the hiPSCs and their derivatives over the course of the protocol, as well as the changes in marker expression that characterize each stage (juxtaposed to the respective photomicrograph). See text for further details. (B) hiPSC colonies on DIV 0. (C) EBs are prepared manually on DIV 1. (D) Nearly all EBs acquire a cystic morphology by DIV 5. (E) One day after plating of the EBs on DIV 6, the cells start to migrate out from the EB. (F) By DIV 11, the hiPSC-derived HFBSCs start to appear and persist until DIV 18. (G) hiPSC-derived keratinocytes form by DIV 25. To obtain engraftable HFBSCs that will yield HFs in vivo following transplantation, the cells should be harvested, as indicated, on DIV 16-18. (Scale bar (A) 30 μm, (B–G) 100 μm). ATRA, all-trans retinoic acid; BMP, bone morphogenic protein; DIV, days in vitro; EBs, embryoid bodies; EGF, epidermal growth factor; hESCs, human embryonic stem cells; HF, hair follicles; HFBSC, hair follicle bulge stem cell; hiPSCs, human induced pluripotent stem cells; hPSC, human pluripotent stem cells; L-AA, L-ascorbic acid.
In Vitro Characterization of hiPSC-Derived HFBSCs (“hiPSC-HFBSCs”)
We performed ICC using primary monoclonal antibodies against KRT18 (an epithelial marker); ITGA6, ITGB1, P63, KRT15, and KRT19 (HFBSCs markers); and KRT14 (a terminally differentiated keratinocyte marker). Also, we monitored the temporal expression of CD200, ITGA6, and ITGB1 in differentiating hiPSCs using FC analysis. Relative gene expression of OCT4, P63, KRT15, KRT19, KRT8, KRT18, KRT5, and KRT14 in hiPSC-derived cells at days in vitro (DIV) 0, 11, 18, and 25 was assessed using qPCR analysis (see Supplemental Material).
Co-Culture of hiPSC-HFBSCs & Mouse Dermal Cells (MDCs) in Vitro
hiPSC-HFBSCs were co-cultured with freshly isolated MDCs using 2 systems: (1) a transwell system, which allows 2 types of cells in monolayer to communicate with each other, but only via diffusible factors because they are separated by a porous membrane that allows the transit of only molecules but not cells; and (2) a 3-dimensional (3D) aggregate of the 2 cell types which allows cell-cell contact. Co-culturing was performed for 1 week, from DIV 11 to DIV 18. For the transwell co-cultures, 2.5x105 hiPSC-HFBSCs were seeded onto a collagen coated surface in the bottom well while an equal number of MDCs were seeded onto permeable transwell inserts in the top well (Corning, Corning, NY). For the 3D co-culture system, equal numbers of hiPSC-HFBSCs and MDCs (2.5x105) were mixed together in a Matrigel supported aggregate (1:1 mixture of Matrigel and “modified FAD medium”) supplemented with BMP-4 (25ng/ml), ATRA (1 μM), and EGF (20 ng/ml). qPCR analysis of hair differentiation markers was performed on the co-cultured hiPSC-HFBSCs at DIV 18. The HF-associated genes assayed included KRT75, msh homeobox 2 (MSX2), lymphoid enhancer-binding factor 1 (LEF1), and trichorhinophalangeal syndrome type I (TRPS1) 23 –27
Transplantation and Characterization of hiPSC-HFBSCs in Vivo
Patch grafting assays were performed as described previously 28,29 . hiPSC-HFBSCs were combined on DIV 16 with freshly isolated MDCs from the backs of black C57BL/6 mice. Equal numbers of HFBSCs and MDCs (2.5 × 106 each) were combined in 100µl phosphate buffered saline (PBS) and injected intradermally into anesthetized (general) SCID mice above the muscle coat, where the cells could remain tightly packed and in contact with each other with little dispersion. Intradermal injection, in contrast to subcutaneous injection, not only prevented dispersion of the cells (cell aggregation and cell-cell interaction proving critical for organogenesis) but also provided a more appropriate microenvironment for the transplanted cells, the dermis being the natural milieu for growing hair.
Three-to-six weeks after implantation, the resulting growths were dissected, and processed for histological and immunohistochemical evaluation (see Supplemental Material). The protocol was approved by the Sanford Burnham Prebys Institutional Animal Care and Use Committee.
Statistical Analysis
One-Way ANOVA was used to calculate the P values.
A P-value of <0.05 was considered significant.
Results
Our goal was to exploit insights from the dynamic up- and down-regulation of the key developmental molecules that determine HF lineage commitment by hPSCs in order to ascertain the precise differentiation stage and molecular requirements—as indicated by surrogate biomarkers—for successful HF replacement in vivo based on the conversion of hiPSCs into engraftable hair generating cells. What emerged from such a study was a strategy that entailed differentiating hiPSCs toward becoming keratinocytes in vitro but, just prior to that end-point, capturing and isolating an intermediate transient progenitor cell population which, we found, expressed HFBSC-associated markers and could generate HFs in vivo before going on to become (less desirable for this purpose) non-HF-producing keratinocytes (Fig. 1). Cells at earlier or later development stages could not yield HFs, suggesting that these intermediate stage cells were bona fide HFBSCs. Although these data are applicable to all hPSCs, we focused on enabling the use of hiPSCs, envisioning that hiPSCs generated from prospective transplant recipients to avoid immunologic incompatibility would have the greatest future clinical use.
Generation and Characterization of hiPSCs
We generated hiPSCs de novo by electroporating, into de-identified normal human skin fibroblasts, non-integrating episomal plasmid vectors encoding OCT3/4, SOX2, KLF4, L-MYC, LIN28 and an shRNA for human p53 18 . The hiPSC clones exhibited appropriate hPSC morphology and were isolated ∼21-45 days post-transfection. Fourteen distinct clones were generated following 2 independent rounds of transfections. Of these 14 clones, 2 unrelated clones were selected at random for the studies described here.
Given that hESCs are the gold standard for hPSCs, hESCs were also studied through all subsequent steps in parallel with the hiPSCs for validation. Like the hESCs, the new hiPSCs expressed the following panel of pluripotency immunomarkers as determined by ICC and FC: the nuclear transcription factors NANOG, SOX2, and OCT4; the surface markers TRA-1-60, TRA-1-81, and SSEA4. Pluripotency of the hiPSC clones was further confirmed by demonstrating their ability to form teratomas containing cells representative of all 3 fundamental primitive germ layers in immunocompromised NSG mice (Supplemental Fig. 1).
Generation, Characterization, & Differentiation of hiPSCs into HFBSCs
With the conversion of hiPSCs into floating EBs (Fig. 1A–C), and the addition of ATRA and L-AA to induce the formation of ectoderm, their expression of classical pluripotency markers began to recede. However, as a bellwether of their ability to ultimately differentiate into HFBSCs, the hiPSCs also expressed CD200, ITGA6, and ITGB1 on their surface -- as confirmed by ICC (Fig. 2A–C; Supplemental Fig. 2) and FC (Fig. 2D–F; Supplemental Fig. 3A–C) —while still co-expressing pluripotency markers (Fig. 2; Supplemental Figs. 2, 3A–C). As detailed in the next section, these HFBSC-associated markers persisted while the pluripotency markers continued to ebb.
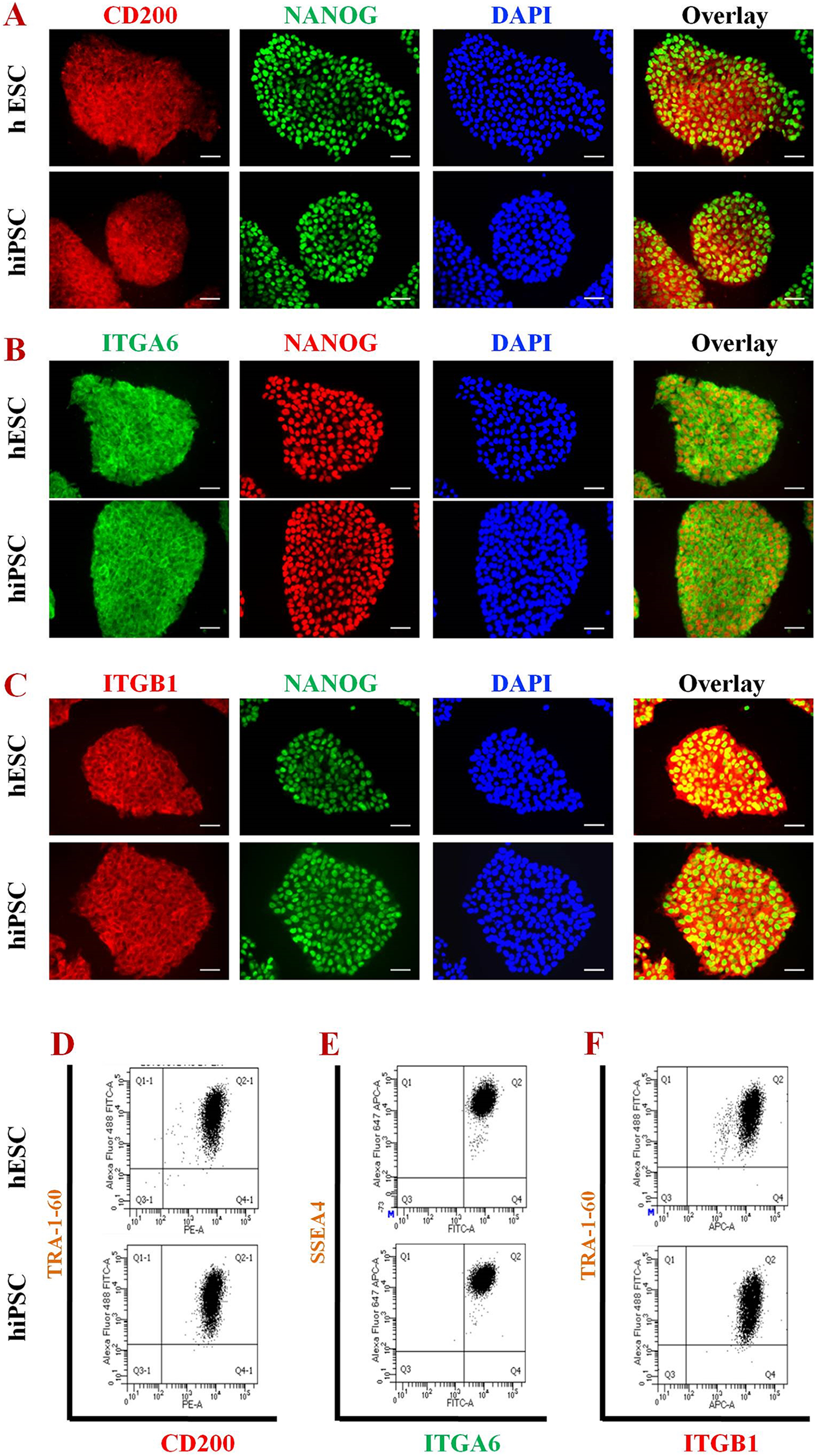
Co-expression of CD200, ITGA6, and ITGB1 along with cardinal pluripotency markers on hESCs and hiPSCs. (A–C) Immunocytochemical (ICC) analysis. (A) CD200 and NANOG; (B) ITGA6 and NANOG; (C) ITGB1 and NANOG. Nuclei of all cells stained blue with DAPI. (Scale bar, 30 μm). (D–F) Flow cytometric (FC) analysis. (D) CD200 and TRA-1-60; (E) ITGA6 and SSEA4; (F) ITGB1 and TRA-1-60. hESCs, human embryonic stem cells; hiPSCs, human induced pluripotent stem cells; ITGA6, integrin α-6; ITGB1, integrin β-1.
By DIV 5, the EBs acquired a cystic morphology, at which time they were plated (Fig. 1D). One day after plating, cells began to migrate out from the plated EBs (Fig. 1E). The addition of BMP-4 precluded the further differentiation of ectoderm into neuroectoderm. EGF enabled growth. Epithelial colonies, which proved to be HFBSCs (as detailed in the next section), started to appear by DIV 11 and persisted until DIV 18 (Fig. 1F); keratinocytes appeared on DIV 25 (Fig. 1G). HFBSCs were harvested no later than DIV 18. Beyond that point, non-engraftable and non-HF-generating keratinocytes would start to emerge.
Expression Dynamics of the Molecules Determining hiPSC-HFBSC Fate
The hPSC-derived cells obtained by the differentiation protocol we devised can be divided into 3 stages: hPSCs (Stage #1) becoming HFBSCs (Stage #2), and then, if no additional cues are presented (including cues from an in vivo environment following transplantation), progression to becoming mature keratinocytes (Stage #3).
Emergence of the expression of KRT8, KRT18, P63, KRT15, and KRT19 indicated differentiation of the hPSCs toward HFBSCs (Fig. 3). Critical for consummation of HFBSC generation was the continued co-expression in these cells of integrins α6 and β1 and the surface glycoprotein CD200 (Fig. 3A–F).
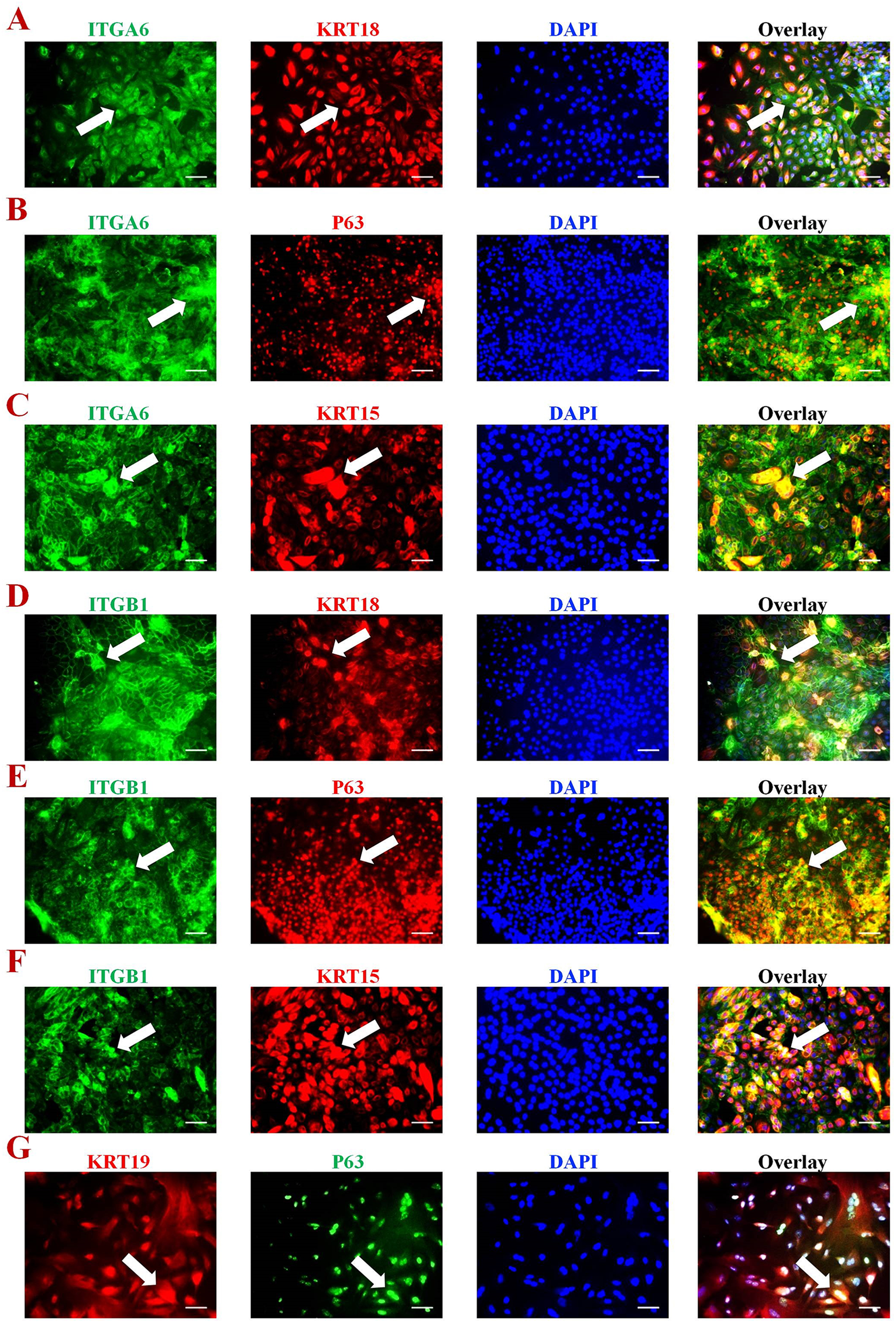
Immunocytochemical characterization of hiPSC-HFBSCs. hiPSC-HFBSCs, as generated by the protocol in Fig. 1, were immunoreactive for (A) ITGA6 and KRT18, (B) ITGA6 and P63, (C) ITGA6 and KRT15, (D) ITGB1 and KRT18, (E) ITGB1 and P63, (F) ITGB1 and KRT15, and (G) KRT19 and P63. Blue nuclear staining with DAPI is used to show all cells in the field. White arrows indicate dual positive cells. ITGA6 and ITGB1 are integrins; KRT18 indicates epithelium; P63, KRT19, and KRT15 are HFBSC markers. The secondary antibody against ITGA6 or ITGB1 is conjugated with FITC (green) and against KRT18, P63 and KRT15 with PE (red) in A-F. In G, the secondary antibody against P63 is conjugated with FITC (green) and against KRT19 with PE (red) (Scale bar, 30 μm). HFBSC, hair follicle bulge stem cell; hiPSC, human induced pluripotent stem cells; ITGA6, integrin α6; ITGB1, integrin β1; KRT, keratin.
In the following paragraphs, we detail on the dynamic changes in these various lineage-determining molecules based on ICC, FC, and q-PCR.
By ICC and FC, expression of the pluripotency markers that defined the hPSCs decreased (Supplemental Fig. 3D, E) concomitant with the emergence of HFBSCs markers (Fig. 3). This was confirmed by RT-PCR; compared to their relative expressions on DIV 0, the expression of OCT4 decreased significantly at DIV 11-18 while the expression of KRT15, KRT19, KRT8, and KRT18 increased significantly at DIV 11-18. These changes heralded the transition from hiPSCs (Stage #1) to hiPSC-HFBSCs (Stage #2). After DIV 18, the cells continued to transition: Compared to their expression on DIV 18, the expression of KRT8 (P < 0.01), KRT18 (P < 0.01), and KRT15 (P < 0.01) decreased significantly by DIV 25, while the expression of KRT5 and KRT14 (keratinocyte markers) increased significantly at DIV 25 (P < 0.01), indicating that the cells had moved past Stage #2 into Stage #3, that of mature -- and potentially terminally differentiated—keratinocytes, a cell type which cannot yield HFs (Fig. 4).
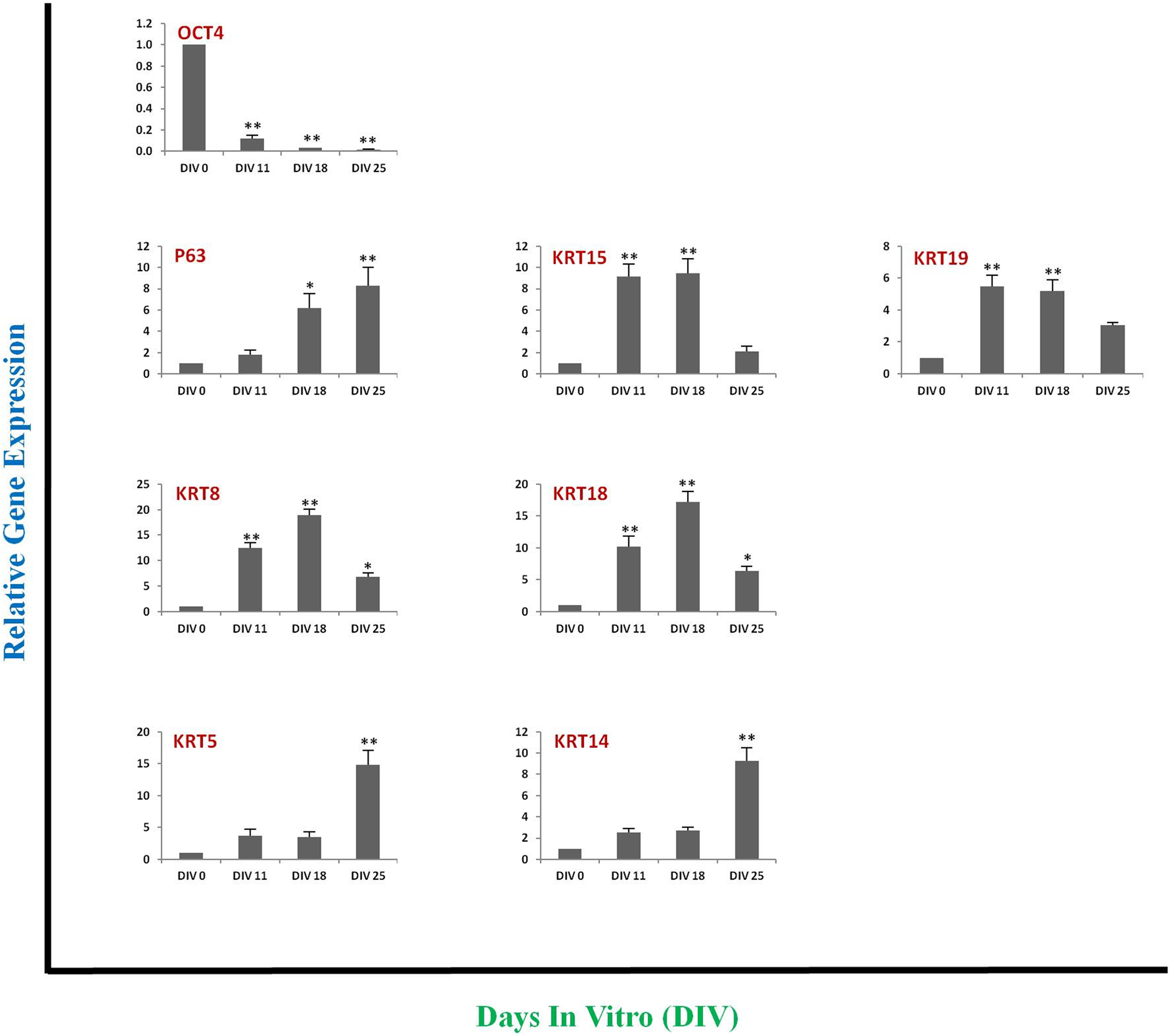
Analysis of the dynamics of the relative temporal expression of molecules associated with pluripotency (OCT4), with HFBSC (P63, KRT15, KRT19, KRT8, KRT18), and with keratinocytes (KRT5, KRT14) in hiPSC-derived cells at various days of the differentiation protocol in Fig. 1. Using RT-PCR, we observed that the expression of OCT4 decreased significantly by DIV 11 and was barely detectable by DIV 18 and DIV 25 compared with its expression at DIV 0. Conversely, the expression of KRT15, KRT19, KRT8, and KRT18 increased significantly at DIV 11-18 compared with their expression at DIV 0. The expression of KRT8, KRT18, and KRT15decreased significantly at DIV 25 compared with their expression at DIV 18 (P < 0.01) while the expression of the keratinocyte markers KRT5 and KRT14 increased significantly at DIV 25 compared with their expression at DIV 0 indicating terminal differentiation (P < 0.01). Data shown are mean ± SD of gene expression from three independent experiments. One-Way ANOVA was used to calculate the P values. * P < 0.05; ** P < 0.01. DIV, days in vitro; hiPSC, human induced pluripotent stem cells; KRT, keratin.
With regard to the surface glycoprotein CD200, its peak expression appeared obligatory for HFBSC generation. CD200 was uniformly expressed by undifferentiated hPSC and continued to be expressed during ectodermal differentiation and differentiation of those ectodermal cells into HFBSCs -- until DIV 18. CD200 expression persisted even as that of the pluripotency markers disappeared during the earliest differentiation stages. However, starting at DIV 18 and reaching a nadir at DIV 25 (Fig. 5A, B), CD200 began to downregulate (∼ 40% of cells have lost CD200 expression by DIV 25) concomitant with increased expression of KRT14 (Fig. 5C), again indicating that the differentiating cells had “cascaded” through and past the HFBSC state (Stage #2) and had entered the state of being mature terminally differentiated keratinocytes (Stage #3), a non HF-generating cell type.
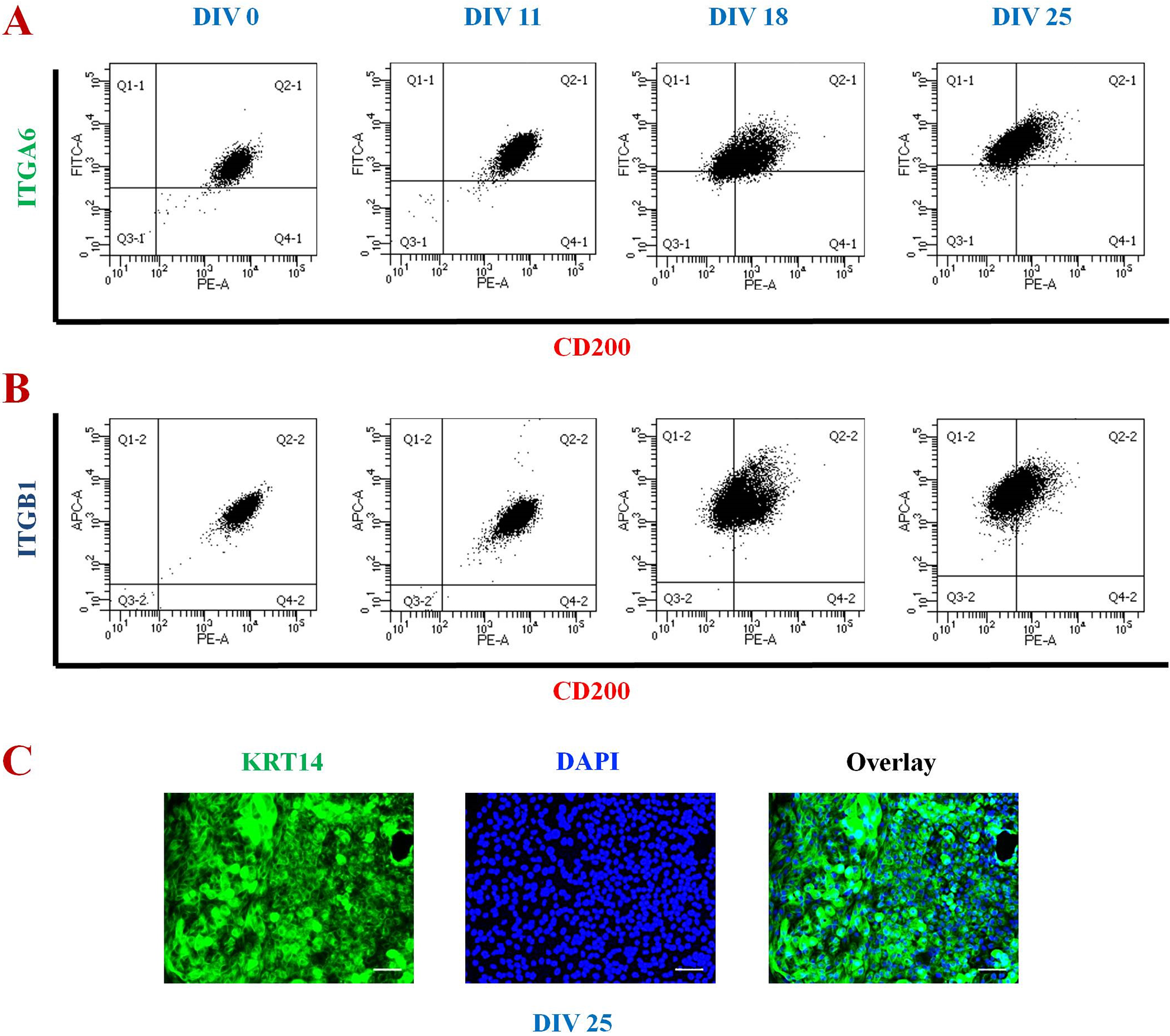
Characterization of the hiPSC-derived keratinocytes in relation to the hiPSC-HFBSCs which emerge earlier. (A, B) Flow cytometric (FC) analysis showing co-expression of (A) CD200 and ITGA6 and (B) CD200 and ITGB1 on hiPSCs on DIV 0-25. In the analyses shown, the upper right quadrant contains cells positive for both ITGA6 and CD200 or ITGB1 and CD200, respectively. The upper left quadrant contains cells that are positive only for ITGA6 or ITGB1. The lower right quadrant contains cells that are positive only for CD200. The lower left quadrant contains double-negative cells. Cells in the upper half are ITGA6+ or ITGB1+ while cells in the lower half are ITGA6– or ITGB1–. Cells on the right side are CD200+ while those on the left side are CD200–. On DIV 0 all of the cells are present at the upper right quadrant, indicating that all of the cells express both ITGA6 and CD200 as well as ITGB1 and CD200. ITGA6 and ITGB1 continue to be expressed on hiPSC-HFBSCs and keratinocytes (i.e., are in the upper right quadrant) from DIV 0 to DIV 25. On DIV18 one can see ∼25% of the cells moving from the upper right quadrant to the upper left quadrant, that is, starting to lose CD200 expression. By DIV 25, more cells have lost CD200 expression and moved from the upper right quadrant to the upper left quadrant (∼40% of the cells); CD200 expression reaches its nadir at this point (DIV 25). It is those CD200– cells that become KRT14+ and KRT5+ mature differentiated keratinocytes on DIV 25 (as illustrated in (C)). (C) Immunocytochemical analysis showing expression of KRT14 at DIV 25 of the differentiation protocol (the point at which CD200 expression has ebbed) indicating the emergence of mature keratinocytes (Stage #3 cells), a point beyond the HFBSC stage (Stage #2 cells) and one that cannot yield HF generation in vivo following transplantation. Blue nuclear staining with DAPI shows all cells in the field. (Scale bar, 30 μm). DIV, days in vitro; HFBSC, hair follicle bulge stem cell; hiPSC, human induced pluripotent stem cells; ITGA6, integrin α6; ITGB1, integrin β1; KRT, keratin.
Hence, downregulation of CD200 with concomitant upregulation of KRT14 appeared to separate the HFBSC stage (Stage #2) from the keratinocyte stage (Stage #3). This “border zone”, we realized, held translational significance if our speculation was supported that Stage #2 cells (hiPSC-HFBSCs) but not Stage 3 cells (KRT14+ keratinocytes), could engraft to yield HFs in vivo.
While that speculation was tested directly via the transplantation studies reported below, we first needed to determine whether Stage 2 cells had the competence to express hair differentiation markers when exposed to proper inductive cues (Fig. 6).
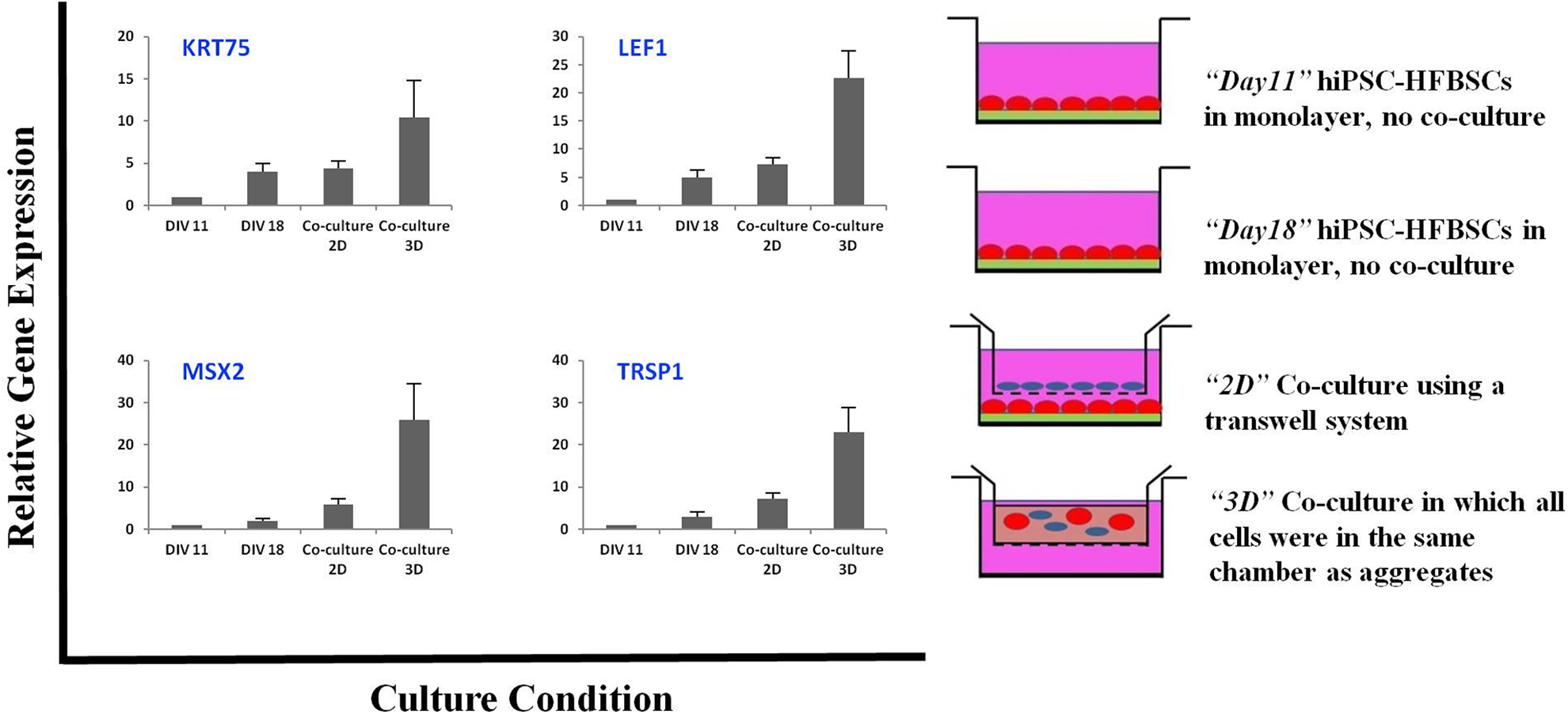
Co-culture of hiPSC-HFBSCs and MDCs. Marked upregulation of hair-related gene expression in hiPSC-HFBSCs following their induction by trichogenic mesenchymal cells via cell-cell contact in 3D co-cultures compared with hiPSC-HFBSC cells alone in monolayer (at DIV 11 or DIV 18), or co-cultures in a transwell system (which allowed interaction between hiPSC-HFBSC cells and mesenchymal cells via diffusible factors alone, the cells separated by a porous membrane that permitted passage of molecules but not cells.) Data shown are mean ± SD of gene expression from three independent experiments. HFBSC, hair follicle bulge stem cell; hiPSC, human induced pluripotent stem cells; KRT, keratin; LEF1, lymphoid enhancer-binding factor 1; MDC, mouse dermal cells; MSX2, msh homeobox 2; TRPS1, trichorhinophalangeal syndrome type I.
The “bulge activation hypothesis” holds that signals from the dermal papillae (DP) (the mesenchymal component of the HF) stimulate resting HFBSCs to generate transient amplifying cells which can then form HFs and hair shafts in vivo 30 . Therefore, we co-cultured hiPSC-HFBSCs with freshly isolated MDCs for 1 week using two different paradigms (Fig. 6). In the first, a transwell system was employed in which hiPSC-HFBSCs were placed as a monolayer in the bottom well while an equal number of MDCs were seeded as a monolayer onto porous inserts in the upper well; the pores were a size that would allow the passage of only molecules but not cells. This system would determine whether the Stage 2 cells in monolayer (2 dimensions [2D]) would respond to diffusible signals. In the second system, equal numbers of the two cell types (hiPSC-HFBSCs and MDCs) were co-cultured in a 3 dimensional (3D) sphere (in which all cells were in the same top chamber as aggregates) allowing for cell-cell contact. qPCR analysis for hair differentiation markers was then performed on the co-cultured hiPSC-HFBSCs (Stage 2 cells) on DIV 18. Not only was HF-associated gene expression in the two co-culture systems compared with each other but also with expression by the Stage 2 cells cultured alone in monolayer on DIV 11 and DIV 18. Expression of the HF-associated genes KRT75, MSX2, LEF1 and TRPS1 showed a marked increase in the 3D co-culture system, much greater than that in the 2D transwell system or in the Stage 2 cells alone on DIV 11 and DIV 18 (Fig. 6). Stated another way, Stage 2 cells exposed to diffusible factors alone for induction (i.e., via transwell) showed an expression of HF-associated genes that was slightly higher than if the cells had been cultured and matured alone in monolayer. These data not only spoke to the likelihood of successful HF generation following engraftment of Stage 2 cells but also the necessity for an in vivo environment with cell-contact in a proper 3D niche to achieve full induction. Indeed, the competence of Stage 2 cells could be missed without transplantation.
Characterization in vivo of hiPSC-HFBSCs Following Transplantation
To confirm our speculation that the optimal molecular profile for inducing hiPSCs to yield HF-competent HFBSCs was the sustained co-expression of CD200, ITGA6 and ITGB1 together with emergence of other HFBSC-associated gene products such as P63, KRT15, KRT19, and KRT18 (i.e., Stage 2 cells), we transplanted hiPSC-HFBSCs into SCID mice at DIV 16. DIV 16 represented the time point at which there was maximal expression of CD200 (before its downregulation, as shown by FC) as well as of KRT15, KRT19, and KRT18 (as shown by RT-PCR). Transplantation of the hiPSC-HFBSCs was accomplished by co-injection of HFBSCs and MDCs intradermally above the muscle coat, where the cells could remain tightly packed and in contact with each other with little dispersion. The hiPSC-derived HFBSCs yielded HF in vivo (Fig. 7). On the other hand, transplantation of cells at DIV 25 -- after downregulation of CD200 -- failed to generate HFs in vivo. In other words, mature keratinocytes, Stage 3 cells, were not competent to generate HFs in vivo. As suggested by the above-described 3D co-culture experiments, cell aggregation and cell-cell interaction were pivotal for the Stage 2 cells to participate in organogenesis and yield HFs. Highly dispersed grafts injected in the subcutaneous fat did not develop HF, nor did injection of MDCs alone.
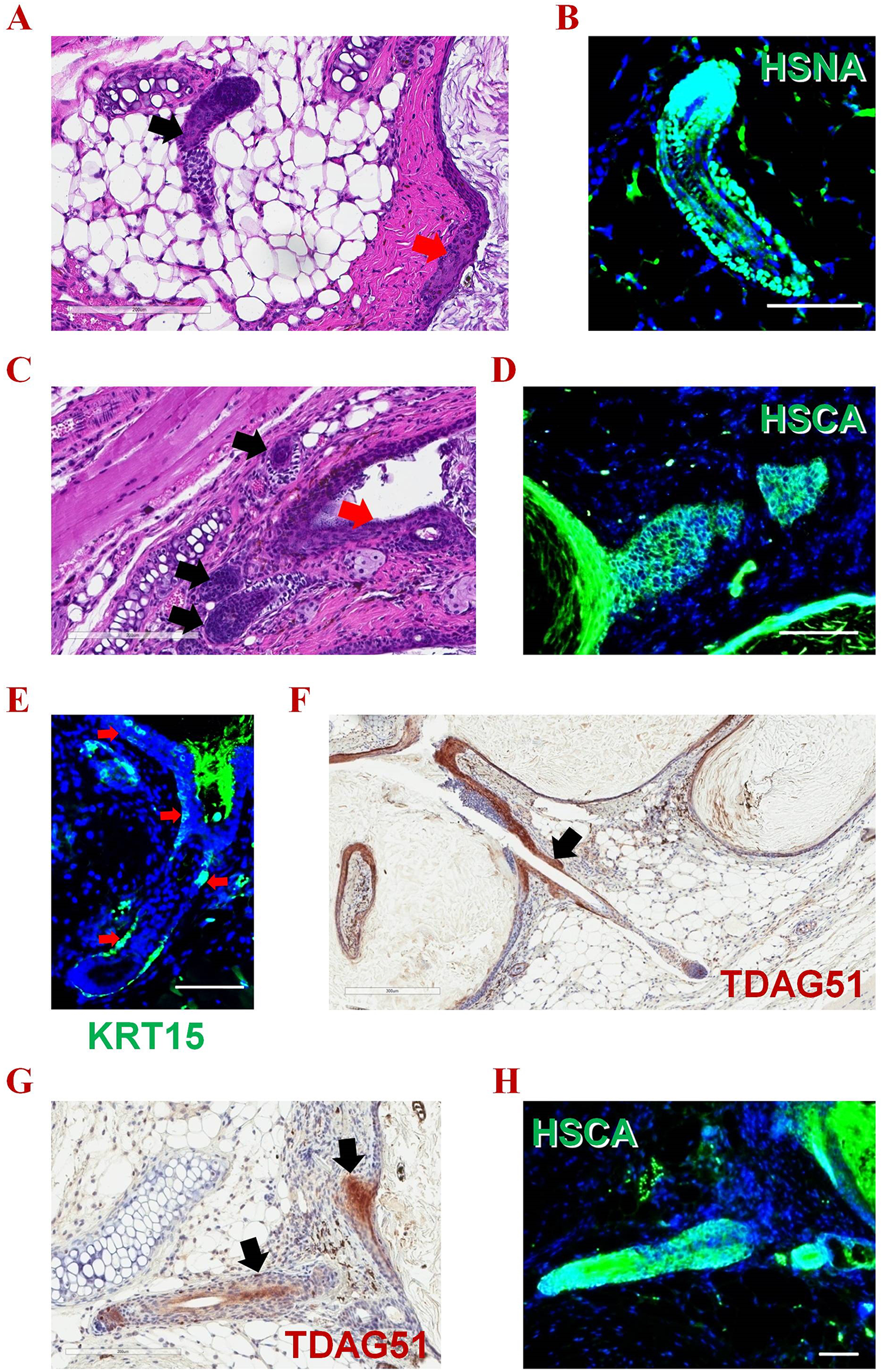
Histologic evaluation of donor-derived HFs following intradermal transplantation of hiPSCs-HFBSCs. Injection of CD200+/ITGA6+/ITGB1+ hiPSC-derivatives intradermally into SCID mice (above the muscle coat such that they can maintain cell-cell contact with little dispersion) resulting in small epidermal cysts with HFs radiating from them, proof of HFBSC differentiation and HF generation competence. (A) A representative HF (black arrow) and epidermal cyst lined by multilayered epidermis (red arrow). (B) Positive immunoreactivity of this HF for HSNA (green). (C) A small epidermal cyst showing a multi-layered epidermis (red arrow) with multiple HFs radiating from it (black arrows). (D) Positive immunoreactivity for HSCA (green) in the reconstituted epidermis and HF. (E) Positive immunostaining (green) of a representative reconstituted HF using an antibody against KRT15 (a known HFBSC stem cell marker); the immunopositive cells are present in the basal layer of epidermis, in the bulge region, and in the basal layer of the outer root sheath (red arrows). (F, G) Positive immunoperoxidase staining (brown) of representative reconstituted HFs with an antibody against TDAG51 (a known HF stem cell marker); the stained cells are present in the bulge region (black arrows). (H) The reconstituted epidermis and HF in (G) is immunopositive for HSCA (green). (Scale bar, 100 μm). (see Supplemental Figs. 4 and 5 for additional immunohistochemistry supporting human origin of the HFs). HF, hair follicle; HFBSC, hair follicle bulge stem cell; hiPSC, human induced pluripotent stem cells; HSCA, human specific cytoplasmic antigen; HSNA, human specific nuclear antigen; ITGA6, integrin α6; ITGB1, integrin β1; KRT, keratin; TDAG51, T-Cell Death-Associated Gene 51.
Three to six weeks following transplantation, histological analysis showed that the injected Stage 2 cells aggregated to form small cystic spheres in the host dermis. Human-like multilayered epidermis was also formed in the grafts. The cysts consisted of both basal keratinocytes and stratified epidermis. Pilosebaceous units growing outward from the cyst were evident (Fig. 7). In Fig. 7, one can see the morphology of the hiPSC-derived HFs, including their discrete component parts (e.g., the hair shaft, the hair matrix, the outer root sheath.). Although the new follicles in this system do not usually produce skin surface hair shafts (because the new follicle growth occurs in the deep dermis), we expected that, if the trichogenic cells were implanted superficially enough, the hair shafts would egress, individually or in tufts. Indeed, the hiPSC-HFBSCs not only reconstituted the epithelial components of the HF but also the interfollicular epidermis. These findings indicated that hiPSC-derived HFBSC were capable of generating epidermis and that they responded to inductive dermal signals in vivo in the developmentally appropriate niche to generate HFs.
Human origin of the epithelial cells in the HFs and epidermis was confirmed by immunopositivity for the human-specific nuclear antigen (HSNA) which was detected in ∼60-70% of growing HFs cells. Human derivation of the cells was further confirmed by the positive immunoreactivity for a human-specific cytoplasmic antigen (HSCA) (Fig. 7B, D, H; Supplemental Figs. 4, 5). Human cells were detected in the generated HFs for at least 6 weeks following transplantation (when the experiments were terminated). Hence, most of the cells comprising the HFs arise from the implanted human-origin cells. That 30-40% of the HF cells were of non-human host origin offered the interesting observation that, in the context of transplantation, endogenous cells also contributed to the epithelial and dermal lineages. The HFs formed in our system do not make contact with host epithelium (neither skin appendages nor epidermis); therefore the host contribution to the epithelium came from the surrounding mesenchyme or circulating cells, a process that implied a mesenchymal-to-epithelial transition 31 . This observation was consistent with earlier reports that either vicinal 32 or bone marrow-derived circulating cells 33 will incorporate into regenerating skin and hair, but suggests that transplantation (including of human cells) can evoke that response from the host.
One of the challenges to be addressed by any cell-based treatment for alopecia is to ascertain that newly generated HFs contain a mechanism for cycling? In other words, do they contain HFBSCs in the bulge region? Therefore, we determined whether the donor-derived HFs bore a “bulge region” in vivo rendering them capable of cycling and renewal. Indeed, we found KRT15 expression in the bulge region of the chimeric HFs and in the basal layer of the epidermis (Fig. 7E). The expression of T-Cell Death-Associated Gene 51 (TDAG51) (also called PHLDA1, Pleckstrin Homology Like Domain Family A Member 1), a HF stem cell marker 34,35 , further confirmed the presence of HFBSCs in the bulge region of the donor-derived HFs (Fig. 7F, G). Never noted were neoplastic cells, cells inappropriate to the dermis, or cell overgrowth or deformation.
Assessing the Necessary & Sufficient Elements for HF Generation
We recognized that an important step in confirming the necessity and causality of our putative suite of developmental determinants was to first eliminate and then re-add each factor systematically and demonstrate first an inability and then a reacquisition of the ability to yield engraftable HF-generating HFBSCs. However, all of the seven molecules (markers) required for complete HF generation were also fundamental to the earliest stages of normal development. We could not knock-out any one of the seven, and certainly not all seven, (even conditionally) in the hPSCs without disrupting normal development and confounding interpretation of the experiments. Furthermore, we could not knock them out even in the starting fibroblasts prior to being reprogrammed into hiPSCs because each is obligatory for the reprogramming process itself as well as for subsequent expansion, self-renewal, and acquisition of pluripotency 36,37 . Given these limitations, we performed a series of studies that would serve the same purpose as a “knock-out” but without perturbing the system and obfuscating interpretation through ambiguity, as detailed in the paragraphs below.
We had mapped the expression profile of each of the markers that we knew hPSC-derived HFBSC ultimately achieve. We could do test transplants of cells at each epoch along the developmental trajectory from “epiblast” (i.e., undifferentiated hPSC) to “ectoderm” to “HFBSC” to “keratinocyte”. Each period had a different constellation of markers among the seven; which constellation, we asked, yielded HFs in vivo?
To reprise the dynamic we reported above, P63, KRT15, KRT18, and KRT19 start to be expressed at DIV 11, peak at DIV18, and decrease by DIV 25. CD200, while expressed earlier than DIV 11 (when the cells are still in their pluripotent state), ebb by DIV 25, the point at which KRT5 and KRT14 expression becomes ascendant, heralding the emergence of keratinocytes. ITGA6 and ITGB1 are expressed starting at DIV 0 through DIV 25; if these integrins are not expressed, HF also fails to develop. Transplantation of cells prior to DIV 11 or after DIV 25 fails to yield HF. Therefore, we could conclude that HF generation requires the expression of CD200, ITGA6, ITGB1, KRT15, KRT18, KRT19, and P63 and that a constellation lacking one or more of these molecules was insufficient to yield HFs.
Therefore, we favored transplantation at DIV 16-18, at which time the cells have lost expression of pluripotency markers, continue to express integrins ITGA6 and ITGB1, have attained optimal expression levels of the HFBSC markers KRT15 and KRT19 (in conjunction with P63), express the epithelial marker KRT18, have not yet experienced downregulation of the surface glycoprotein CD200, have not yet started expressing the keratinocyte-associated molecules KRT5 and KRT14, and have attained optimal proliferative capacity to allow a well-populated graft. The optimal site for transplantation was intradermal and above the muscle coat to prevent cell dispersion. Transplantation of MDCs alone failed to yield HFs in vivo.
Discussion
There has long been a debate over how best to produce and select pluripotent stem cell derivatives for reliable and efficient therapeutic HF replacement. We took a developmental approach to this question by generating HFs as they might emerge if starting in the epiblast (a stage emulated by hESCs and, for clinical use, patient-specific hiPSCs) and progressing iteratively through gastrulation and on to dermal organogenesis.
We first mapped the dynamic up- and down-regulation of specific molecules that serve as landmarks for each stage of this developmental process. We pinpointed the precise point at which the appropriate molecular and developmental requirements were attained by the cell (as indicated by surrogate biomarkers) to yield optimal HF generation in vivo if transplanted. What emerged was a 3-stage profile that entailed differentiating hiPSCs (Stage #1) toward becoming keratinocytes in vitro (Stage 3) but, just prior to that end-point, capturing and isolating an intermediate transient progenitor cell population (Stage #2), which we found to be bona fide HFBSCs, not only based on their co-expression of HFBSC-associated molecules but, most importantly, by their ability to generate HFs in vivo upon transplantation. Cells that cascaded beyond “HFBSC Stage #2” into “keratinocyte Stage #3” lost that ability to generate HFs upon grafting. In short, to ensure that hiPSC-derived HFBSC have acquired the competence for HF generation, they must come to express, at the time of transplantation, CD200 10,11 , ITGA6 4,5 , and ITGB1 6 –9 on their surface, and KRT18 12 , P63 5,13 , KRT15 5,14 , and KRT19 5,15,16 , but not KRT5 or KRT14 (keratinocyte markers), intracellularly. Expression of the HFBSCs markers P63, KRT15, and KRT19 as well as the HFBSC-associated epithelial marker KRT18 increases significantly from DIV 11 until DIV 18 (marking the transition from hiPSC Stage #1 to HFBSC Stage #2. Although CD200 is expressed starting in Stage #1, it begins to wane at DIV 18, reaching its nadir at DIV 25. Keratinocyte markers KRT5 and KRT14 are low through Stages #1 and #2, but dominate by DIV 25. DIV 18 appears to represent a developmental border between Stage #2 (CD200+ engraftable HF-yielding HFBSCs) and Stage #3 (unengraftable mature non-HF yielding KRT5+/14+ keratinocytes). Therefore, to yield HFs, transplantation, we learned, should take place after DIV 11 but no later than DIV 18; we have chosen DIV 16-18 as our optimal transplant time to insure that all pluripotency markers have downregulated and the HFBSC-associated molecules are at their peaks.
Our observation that expression of CD200, ITGA6 and ITGB1 on hiPSCs and hESCs is a sine qua non for the differentiation of hPSCs into HFBSCs has, heretofore, been surprisingly controversial in the literature. The expression of these markers on hPSCs has been variously reported as present or absent over the years by past investigators 3,17,38 –40 . Our ICC and FC data not only clearly confirm their expression on Stage #1 cells, in association with other known pluripotency markers such as NANOG, SOX2, TRA-1-60, TRA-1-81, and SSEA4, but we further speculate that, in the absence of such integrin-related molecules and glycoproteins, hPSCs cannot proceed to becoming HFBSC Stage #2 cells with competence for yielding HFs. Evidence in support of that conjecture comes from two observations: First, we witnessed in our 3D co-cultures that induction of the molecules mediating HF generation required cell-cell contact between receptive HFBSCs and trichogenic mesenchymal cells, an interaction mediated by these integrins 41 . Second, downregulation of CD200 coincided with progression of Stage #2 HFBSCs toward a Stage #3 keratinocyte fate incapable of HF generation, while transplantation of the Stage #2 cells at the peak of their CD200 expression in conjunction with expression of the integrins consistently yielded HFs in vivo.
Beyond our empiric data, the expression of these surface glycoproteins makes biologic sense for creating hPSC-derived HFs. Integrins are transmembrane glycoproteins composed of an α subunit and a β subunit that are linked via non-covalent bonds. The α6 subunit associates with the β1 subunit or the β4 subunit to form α6β1 and α6β4 integrin heterodimers. α6β1 is expressed on a variety of cell types and functions as a cellular receptor for matrix laminin 42 . ITGA6 is the α6 subunit (also known as CD49f); ITGB1 is the β1 subunit. α6β1 integrins along with α6β4 integrins confer functional characteristics to stem cells 43 ; α6β1 may be the dominant integrin 37 . More than 30 different stem cell types have been found to express ITGA6 on their membranes 36,44 . During the reprogramming of fibroblasts into hiPSCs, ITGA6 is upregulated and focal adhesion kinase (FAK) is inactivated (via dephosphorylation). During differentiation of the hiPSCs, the converse takes place: ITGA6 levels diminish and FAK is activated (phosphorylated at residue Y397). Activation of ITGB1 also leads to FAK phosphorylation and reduction of NANOG, OCT4, and SOX2. Knockdown of ITGA6 can mimic ITGB1 activation and reduce or eliminate normal hESC and hiPSC colony development, self-renewal, and pluripotency 37 . Hence, while the presence of these two arms of the integrin system are necessary for hPSC maintenance and differentiation 36 , they may play dynamic opposing roles; obviously an equilibrium must be struck between them for first reprogramming and then development to proceed -- in this case, all the way to becoming HF-competent HFBSCs. CD200 is a glycoprotein widely expressed on cancer stem cells (breast, colon, hematopoietic) and may be required to maintain growth, self-renew, metastasis, and to evade the immune system 38 . It is also a marker of HFBSCs 11 (as well as limbal stem cells 45 ). This combination of surface molecules likely interacts with specific matrix receptors during culture which, in turn, influences the hiPSCs’ adhesion, survival, proliferation, and entrance into particular differentiation “programs” 17 . In our study, the expression of these molecules on hiPSCs appeared to ensure that the cells had acquired the molecular competence for becoming HFs in vivo. In fact, the necessity of these surface markers may be used for cytometry to extract hiPSC derivatives in Stage #2 that can yield HFs from a heterogeneous population that might contain cells disadvantageous for this therapeutic indication. For regulatory purposes, it might be a way to isolate therapeutic Stage #2 cells from undifferentiated and potentially tumorigenic hPSCs. These same markers, which are avatars for a desirable developmental trajectory, may be used to identify drugs that might enhance the efficiency of HF generation.
In conclusion, based on the developmental dynamics mapped above, we favor transplantation at DIV 16-18, at which time the cells have lost expression of pluripotency markers, continue to express integrins ITGA6 and ITGB1, have attained optimal expression levels of the HFBSC-associated markers KRT15, KRT18, and KRT19 (as well as P63), have not yet experienced downregulation of CD200, have not yet started expressing the keratinocyte-associated molecules KRT5 and KRT14, and have attained optimal proliferative capacity to allow a well-populated graft. Transplantation should be done intradermally above the muscle coat, where the cells can remain tightly packed and in contact with each other with little dispersion. The guidance provided by the present study may bring us closer to replacing HFs lost in non-cicatricial and cicatricial alopecia through the use of a patient’s own hiPSC-derived follicular stem cells.
Supplemental Material
Supplemental Material, sj-pdf-1-cll-10.1177_09636897211014820 - The Developmental & Molecular Requirements for Ensuring that Human Pluripotent Stem Cell-Derived Hair Follicle Bulge Stem Cells Have Acquired Competence for Hair Follicle Generation Following Transplantation
Supplemental Material, sj-pdf-1-cll-10.1177_09636897211014820 for The Developmental & Molecular Requirements for Ensuring that Human Pluripotent Stem Cell-Derived Hair Follicle Bulge Stem Cells Have Acquired Competence for Hair Follicle Generation Following Transplantation by Michel R. Ibrahim, Walid Medhat, Hasan El-Fakahany, Hamza Abdel-Raouf and Evan Y. Snyder in Cell Transplantation
Footnotes
Abbreviations used
Acknowledgements
We thank the Egyptian Ministry of Higher Education and Scientific Research for funding this research. We thank Yoav Altman and the SBP Flow Cytometry Core for help in performing and analyzing experiments on the Image Stream Imaging Flow Cytometer. We thank Donglin Lao and Yang Liu of the Sanford Burnham Prebys Medical Discovery Institute for their excellent technical assistance. Also, we thank Sandra L. Leibel and Rachael McVicar for their help in the 3D culture system.
Authors’ Contributions
Michel R. Ibrahim was responsible for conception; study design; data collection, analysis, and interpretation; and manuscript writing. Walid Medhat, Hasan El-Fakahany, and Hamza Abdel-Raouf were responsible for data collection, assembly, and analysis. Evan Y. Snyder was responsible for overall supervision, conception and design, data analysis and interpretation, financial support, manuscript writing, and final approval of the manuscript.
Ethical Approval
The hiPSCs were established from archived fibroblasts obtained from anonymous donors, hence falling under NIH Exemption #4. The use of all human pluripotent stem cell lines was approved by the Sanford Burnham Prebys Institutional Review Board (IRB) and Stem Cell Research Oversight Committee (SCRO).
Statement of Human and Animal Rights
All transplantation was approved by the Sanford Burnham Prebys Institutional Animal Use and Care Committee (IACUC protocol number “AUF” 16-040)
Statement of Informed Consent
No human subjects were used in this work; therefore, informed consent was not applicable. The fibroblasts used to establish the hiPSCs were obtained from a bank of de-identified donors.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article or its supplemental materials
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was funded by the Egyptian Ministry of Higher Education and Scientific research.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.