
Abstract
While neural stem cells (NSCs) are widely expected to become a therapeutic agent for treatment of severe injuries to the central nervous system (CNS), currently there are only few detailed preclinical studies linking cell fate with experimental outcome. In this study, we aimed to validate whether IV administration of allogeneic NSC can improve experimental autoimmune encephalomyelitis (EAE), a well-established animal model for human multiple sclerosis (MS). For this, we cultured adherently growing luciferase-expressing NSCs (NSC-Luc), which displayed a uniform morphology and expression profile of membrane and intracellular markers, and which displayed an in vitro differentiation potential into neurons and astrocytes. Following labeling with green fluorescent micron-sized iron oxide particles (f-MPIO-labeled NSC-Luc) or lentiviral transduction with the enhanced green fluorescent protein (eGFP) reporter gene (NSC-Luc/eGFP), cell implantation experiments demonstrated the intrinsic survival capacity of adherently cultured NSC in the CNS of syngeneic mice, as analyzed by real-time bioluminescence imaging (BLI), magnetic resonance imaging (MRI), and histological analysis. Next, EAE was induced in C57BL/6 mice followed by IV administration of NSC-Luc/eGFP at day 7 postinduction with or without daily immunosuppressive therapy (cyclosporine A, CsA). During a follow-up period of 20 days, the observed clinical benefit could be attributed solely to CsA treatment. In addition, histological analysis demonstrated the absence of NSC-Luc/eGFP at sites of neuroinflammation. In order to investigate the absence of therapeutic potential, BLI biodistribution analysis of IV-administered NSC-Luc/eGFP revealed cell retention in lung capillaries as soon as 1-min postinjection, resulting in massive inflammation and apoptosis in lung tissue. In summary, we conclude that IV administration of NSCs currently has limited or no therapeutic potential for neuroinflammatory disease in mice, and presumably also for human MS. However, given the fact that grafted NSCs have an intrinsic survival capacity in the CNS, their therapeutic exploitation should be further investigated, and—in contrast to several other reports—will most likely be highly complex.
Keywords
Introduction
Multiple sclerosis (MS) is generally defined as a chronic autoimmune inflammatory disease of the central nervous system (CNS) and is characterized by inflammatory lesions in spinal cord and brain, which lead to demyelination, gliosis, and eventually to loss of neuronal function (10,50). Experimental autoimmune encephalomyelitis (EAE), the complementary preclinical animal model for MS, is widely used to study the therapeutic and/or regenerative potential of different therapeutic agents and/or cellular therapies (4,21,55). With regard to the latter, neural stem cell (NSC) transplantation has the potential to become a cellular therapy for treatment of severe injuries to the CNS. Several studies have indeed shown the efficacy of NSC transplantation in animal models for the treatment of Parkinson's disease, stroke, and EAE (2,16,20,37,57), although the precise mechanisms of symptomatic improvement following NSC transplantation remain to be elucidated before large-scale human clinical trials can be initiated.
As MS and EAE are heterogeneous diseases, with lesions at various sites throughout the CNS, IV NSC administration has been proposed to be of particular interest due to the intrinsic capacity of NSCs to migrate towards inflammatory lesions (7,26,39). Although several studies reported on a beneficial effect of IV administered autologous NSCs on the clinical course of EAE (37,56), recent reports indicate that most—if not all—IV administered cells do not enter the CNS, but exert their therapeutic effect via immunomodulatory mechanisms throughout the peripheral immune system (6,15,38). Therefore, further studies to link the fate of NSCs in vivo with experimental outcome will be mandatory to understand their regenerative potential. To this end, we here aimed to preclinically validate whether and/or how IV administration of adherently cultured allogeneic NSCs can prevent EAE onset and disease progression. This study therefore distinguishes itself from previously published reports by: 1) the transplantation of a uniform adherently cultured NSC population, which allows for a better characterization of cell graft and function, and 2) the transplantation of allogeneic NSCs, which is of high clinical relevance with regard to potential future human clinical trials.
NSC are traditionally isolated and cultured from embryonic (13,27,53) or adult brain tissue (11,31,43–45), and proliferate in suspension culture as spherical aggregates, designated as neurospheres. These neurospheres consist of multipotent stem cells that are normally present in very small numbers, and progenitor cells that are more restricted in proliferation and differentiation potential (22,36). Novel culture protocols have recently been described in order to expand large numbers of self-renewable uniform populations of adherently growing NSCs from embryonic stem cells and embryonic brain tissue (12,19,40). The latter overcomes specific problems associated with neurosphere cultures (e.g., substantial cell mortality and nonuniformity of the cell populations obtained). Therefore, in the first technologically oriented part of this study, we focus on culture, in vitro characterization, intracerebral transplantation, and in vivo bioimaging of adherently growing NSCs cultured from embryonic mouse brain.
While preclinical development of NSC-based cell therapies in small animal models is mainly using autologous cell grafts, less attention is given to the immunological aspects of allogeneic cell transplantation. The latter will however become of major interest, because—from a practical and financial point of view—future human NSC-based therapies for CNS injuries will most likely be performed using well-characterized (and possibly genetically engineered) allogeneic NSC populations (5,24,42,46,48). The use of such “off-the-shelf” stem cell preparations provides many advantages over the use of autologous cell preparations as they can be premanipulated and/or screened for increased effectiveness (e.g., regenerative capacity) without adverse effects (e.g., tumorigenicity). However, the use of allogeneic cell implants implies the need for immune suppression (e.g., cyclosporine A, CsA) in order to avoid acute or chronic rejection of cellular transplant in the CNS, as previously demonstrated by us (8,47,52) and others (9,28). Therefore, in the second therapeutically oriented part of this study, we focus on the regenerative potential of IV administered allogeneic NSCs on EAE disease progression under immunosuppressive therapy.
Following the above described reasoning, IV administration of allogeneic adherently cultured NSCs in combination with immunosuppressive therapy will most likely be the first option to enroll NSCs into large clinical trials for the treatment of MS. In this context, immune suppressive therapy will have the dual function to inhibit ongoing pathological immune responses (i.e., myelin-specific T-cells) and to prevent immune-mediated rejection of allogeneic NSC administered. On the other hand, administration of NSCs is hoped to contribute to myelin regeneration by itself or to stimulate myelin regeneration by endogenous NSCs. In this study, we performed multiple transplantation experiments using well-defined NSC cultures and aim to provide a detailed report regarding the clinical relevance of IV administration of allogeneic NSCs for treatment of murine EAE in view of the potential application of NSCs for treatment of human MS.
Materials and Methods
Animals
Homozygous ROSA26-L-S-L-Luciferase transgenic mice (49) (FVB background) were obtained from Jackson Laboratories (strain code 005125) and further bred in the animal facility of the University of Antwerp (UA). Male and female embryos (12–16 days postcoitus, n = 8) were used for initiating NSC cultures. Male and female newborn mice (P0, n = 21) were used for initiating primary astrocyte and neuron cultures. Adult females were used for cell implantation experiments (n = 26) or dendritic cell culture (n = 2). Adult wild-type C57BL/6J mice were obtained from Charles River Laboratories (strain code 027). Female mice (n = 161) were used for induction of EAE and/or cell transplantation experiments. For all experiments, mice were kept in normal day-night cycle (12/12) with free access to food and water. All experimental procedures were approved by the Ethics Committee for Animal Experiments of the UA (approval No. 2009/1).
NSC Cultures
NSCs were cultured from embryonic brains of FVB-derived ROSA26-L-S-L-Luciferase transgenic mice following a protocol previously described by Conti et al. (12), with minor modifications. Briefly, embryonic brains (E12–16) of ROSA26-L-S-L-Luciferase transgenic mice were enzymatically dissociated using a 0.2% collagenase A (Roche)/DNase-I (1000 Kunitz units/50 ml, Sigma) solution in phosphate-buffered saline (PBS) for 2 h at 37°C in a shaking water bath. The cell population obtained was then resuspended in 10 ml neural expansion medium (NEM), consisting of Neurobasal A medium (Invitrogen) supplemented with 10 ng/ml epidermal growth factor (EGF, ImmunoTools), 10 ng/ml human fibroblast growth factor-2 (hFGF-2, ImmunoTools), 100 U/ml penicillin (Invitrogen), 100 mg/ml streptomycin (Invitrogen), 0.5 μg/ml amphotericin B (Invitrogen), and 1% modified N2 supplement. The modified N2 supplement consisted of DMEM/F12 medium (Gibco) supplemented with 7.5 mg/ml bovine serum albumin (BSA, Invitrogen), 2.5 mg/ml insulin (Sigma), 2 mg/ml apotransferrin (Sigma), 0.518 μg/ml sodium selenite (Sigma), 1.6 mg/ml putrescine (Sigma), and 2 μg/ml progesterone (Sigma). Cells were plated in a T25 culture flask in order to obtain a neurosphere population. Next, neurospheres were dissociated using accutase (Sigma) and cells were plated in 10 ml NEM on fibronectin-coated (5 μg/ml in PBS, R&D Systems) T25 culture flasks in order to allow outgrowth of adherently growing NSC cultures. Following 24 h of culture, nonadherent cells were removed and 10 ml fresh NEM was added to the cultures. After 7 days, cultured cells were harvested following accutase treatment for 5 min at 37°C and replated in a new fibronectin-coated T25 flask in 10 ml NEM (passage 1). For routine cell culture, NEM was replaced each 3–4 days and NSC cultures were split 1:5 every 7 days. All cultured cells were incubated at 37°C and 5% CO2. For immunofluorescence staining, harvested NSCs were cultured on fibronectin-coated glass cover slips.
In Vitro Differentiation of NSC Cultures
NSCs were harvested using accutase treatment and replated on glass cover slips coated with poly-L-ornithine (Sigma) in a 24-well plate in 500 μl of neural differentiation medium (NDM), consisting of Neurobasal A medium supplemented with Neurocult? Neural Stem Cell Differentiation Supplements (StemCell Technologies). Every 3–4 days half of the NDM was refreshed during a period of 1–3 weeks. Immunofluorescence staining was directly performed on differentiated NSC cultures.
Primary Astrocyte and Neuron Cultures
Mixed primary glial cortical and neuronal cultures were prepared from newborn ROSA26-L-S-L-Luciferase transgenic mice (P0). Briefly, after removal of meninges and blood vessels, cerebral cortices were dissociated mechanically using sterile scissors and treated enzymatically with a 0.2% collagenase A/DNase-I (1000 Kunitz units/50 ml) solution (in PBS) for 90 min at 37°C in a shaking water bath. Afterwards, large tissue pieces were removed by passing through a 100-μm nylon filter (BD Falcon). The cell suspension obtained was then washed twice with PBS and resuspended in DMEM complete medium, consisting of Dulbecco's modified Eagle medium (DMEM, Invitrogen) supplemented with 10% fetal calf serum (FCS, Hyclone), 100 U/ml penicillin, 100 mg/ml streptomycin, and 0.25 μg/ml amphotericin B. For primary astrocyte cultures, the total cell population was plated on poly-L-ornithine-coated glass cover slips in a 24-well plate in DMEM complete medium. For primary neuron cultures, the total cell population was plated on poly-D-lysine-coated (Sigma) glass cover slips in a 24-well plate in Primary Neuron Growth Medium (PNGM™, Lonza). For both cultures, following a first incubation of 24 h, nonadherent cells were removed and medium was refreshed. Every 3–4 days half of the medium was refreshed during a period of 1–3 weeks. Immunofluorescence staining was directly performed on cultured neurons and astrocytes.
Bone Marrow-Derived Stromal Cell Cultures
Bone marrow-derived stromal cells (BMSCs) from ROSA26-L-S-L-Luciferase transgenic were cultured as previously described (8). Briefly, established BMSCs cultured were passaged 1:3 twice a week in complete expansion medium (CEM), consisting of Iscove's modified Dulbecco's medium (IMDM, Cambrex) supplemented with 8% horse serum (HS, Invitrogen), 8% FCS, penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively), and amphotericin B (0.25 μg/ml). For immunofluorescence staining, harvested BMSCs were cultured on fibronectin-coated glass cover slips.
Dendritic Cell Cultures
Dendritic cells (DCs) were cultured from bone marrow (BM) of FVB-derived ROSA26-L-S-L-Luciferase transgenic mice, following a protocol previously described by Lutz et al. (32), with minor modifications. Briefly, BM cells flushed from femurs and tibias of 8–12-week-old female mice were dispersed through a 100-μm nylon cell strainer to obtain a single cell solution. To initiate DC cultures, 5 × 106 BM cells were seeded in a T175 culture flask in 20 ml DC medium with the following composition: RPMI-1640 medium supplemented with L-glutamine (Invitrogen), 10% FCS, 50 μM β-mercaptoethanol, penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively), and 20 ng/ml recombinant mouse granulocyte-macrophage colony-stimulating factor (rmGM-CSF, ImmunoTools). At day 3 of culture, an additional 20 ml of DC medium was added to each culture flask. At days 6 and 8, half of the culture supernatant was collected, centrifuged, and the cell pellet was resuspended in 20 ml of fresh DC medium. Cells were harvested and used for transplantation experiments at day 10 of culture.
Messenger RNA Transfection of NSC Cultures
Messenger (m)RNA encoding the enhanced green fluorescent protein (eGFP) and the Cre recombinase protein was prepared as described previously (41,54). Directly prior to mRNA lipofection, fresh mRNA (5 μg)/Lipofectamine™ 2000 (10 μl) mixtures were prepared in 600 μl OptiMem medium (Invitrogen), according to the manufacturer's instructions. Next, subconfluent (70%) T25 flask cultures of NSCs were washed twice with PBS and fresh NEM without antibiotics (10 ml) + mRNA lipoplexes (600 μl) were added to the cultures. After 4 h, medium was refreshed in order to remove remaining lipoplexes. After 24 h of culture, eGFP mRNA-lipofected NSCs were analyzed for eGFP expression and cell viability by flow cytometric analysis. After 72 h of culture, Cre mRNA-lipofected NSCs were analyzed for luciferase expression using a luciferase assay. Cre mRNA-lipofected NSC cultures were then further expanded as described above and the in vitro luminescence signal was measured at multiple passages.
In Vitro Luminescence Assay
Luciferase activity in Cre mRNA-lipofected NSCs (NSC-Luc, 1 × 105 cells per assay) was measured using the commercial Bright-Glo luciferase assay system (Promega), according to the manufacturer's instructions. The luminescence signal was quantified using the Envision (Wallac) 2103 Multilabel Reader (Perkin Elmer) and expressed as photons per second (ph/s).
Labeling of NSC Cultures with Fluorescent Micron-Sized Iron Oxide Particles (f-MPIO)
For labeling of cultured NSC with f-MPIO (Bangs Laboratories, ME02F), the following procedure was optimized: 40 h before cell harvest, NSC cultures were incubated with 0.96 μm polystyrene particles containing magnetite (Fe3O2) and a fluorophore (Dragon Green) at a concentration of 5 × 106 f-MPIO particles/ml. After 16-h incubation, cells were washed twice with PBS, followed by further incubation for 24 h in NEM.
Lentiviral Vector Construction
Construction of the pCHMWS-eGFP-IRES-Pac plasmid was performed in two consecutive steps using standard cloning techniques. First, the puromycin resistance gene (Pac) was inserted downstream of an IRES element and the resulting IRES-Pac clone was amplified by PCR and cloned after the eGFP in the pCHMWS-eGFP plasmid (3).
Lentiviral Vector Transduction
Lentiviral vector production was performed as described previously by Geraerts et al. (18), with minor modifications. Filtered vector particles were concentrated using Vivaspin 15 columns (Vivascience, Hannover, Germany), aliquoted, and stored at −80°C. For transduction experiments, cells were seeded in a 24-well plate at 25,000 cells per well. The next day, cells were transduced with vector expressing the eGFP-IRES-Pac cassette (2.86 × 105 pg p24/well) in NEM medium. After 48 h of incubation, the cells were washed and medium was replaced with NEM supplemented with 1 μg/ml puromycin (puro, InvivoGen) for 5 days. Next, cells were subcultured at least four times in NEM + puro and transduction efficiency was determined by flow cytometry.
Flow Cytometry
For immunophenotyping of NSC cultures derived from ROSA26-L-S-L-Luciferase transgenic mice, flow cytometric analysis was performed using the following directly labeled monoclonal antibodies: phycoerythrin (PE)-labeled rat anti-mouse ‘stem cell antigen 1” (Sca-1, eBioscience, 12/5981–82) and PE-labeled rat anti-mouse CD45 (Benton Dickinson, 553081). In addition, the following unlabeled monoclonal antibodies were used: mouse anti-mouse A2B5 (Chemicon, MAB312R) in combination with a fluorescein isothiocyanate (FITC)-labeled goat anti-mouse secondary antibody (AbD Serotec, STAR86F) and rat anti-mouse neural cell adhesion molecule (NCAM, Chemicon, MAB310) in combination with a PE-labeled goat anti-rat secondary antibody (Jackson Immunoresearch, 112–116–143). Before antibody staining, harvested NSCs were washed twice with PBS and resuspended in PBS (2 × 106 cells/ml). For antibody staining, 1 μg of each primary antibody was added to 100 μl of cell suspension for 30 min at 4°C. In case of secondary antibody staining, cells stained with unlabelled primary antibody were washed twice with PBS and resuspended in 100 μl PBS with 1 μg of secondary antibody added to the cell suspension for 20 min at 4°C. Next, antibody-labeled cells were washed twice with PBS, resuspended in 1 ml PBS, and analyzed using an Epics XL-MCL analytical flow cytometer (Beckman Coulter). For determination of eGFP transgene expression, harvested eGFP mRNA-lipofected or lentiviral vector-transduced NSC cultures were washed once with PBS, resuspended in PBS, and directly analyzed. For evaluation of f-MPIO labeling of NSC cultures, harvested f-MPIO-labeled NSC cultures were washed once with PBS, resuspended in PBS, and directly analyzed. For all measurements, cell viability was assessed through addition of GelRed (1× final concentration, Biotum) to the cell suspension immediately before flow cytometric analysis. At least 10,000 cells were analyzed per sample and flow cytometry data were analyzed using FlowJo software.
Immunofluorescence Microscopy
Immunofluorescence staining of NSC cultures, differentiated NSC cultures, BMSC cultures, and primary astrocyte and neuron cultures was performed using the following unlabeled antibodies: a polyclonal rabbit anti-glial fibrillary acidic protein (GFAP, Abcam, AB7779) in combination with Texas Red™ (TR)-labeled goat anti-rabbit secondary antibody (Abcam, AB6719), a polyclonal rabbit anti-sex determining region Y-box 2 (SOX2, Chemicon, AB5603) in combination with TR-labeled goat anti-rabbit secondary antibody, a polyclonal rabbit anti-brain lipid binding protein (BLBP, Chemicon, AB9558) in combination with TR-labeled goat anti-rabbit secondary antibody, a monoclonal mouse anti-mouse neuron-specific beta-3 tubulin (Tuj1, R&D Systems, MAB1195) in combination with FITC-labeled goat anti-mouse secondary antibody (Jackson Immunoresearch, 115–095–164), and a monoclonal rat anti-mouse NCAM (Chemicon, MAB310) in combination with Alexa Fluor® 350-labeled goat anti-rat secondary antibody (Invitrogen, A21093). In brief, cell cultures grown on cover slips were washed twice with PBS, followed by fixation with 8% paraformaldehyde in 100 mM HEPES buffer (Invitrogen) for 30 min at room temperature (RT). Next, cover slips were washed in 200 mM HEPES buffer and incubated for 30 min in 0.1% Triton-X in 200 mM HEPES buffer for permeabilization. Cover slips were then incubated at 37°C in 3% BSA, followed by a washing step with PBS and overnight incubation at 4°C with primary antibody. The following day, cover slips were washed with PBS and incubated with secondary antibody for 1 h at 37°C, followed by a washing step and incubation for 10 min at RT with 4′,6-diamidino-2-phenylindole (DAPI). Finally cover slips were washed in PBS and demineralized water, followed by mounting. Visualization of immunostained cells was performed using a standard research fluorescence microscope (Olympus BX51 fluorescence microscope) equipped with an Olympus DP71 digital camera. Olympus Cell-F Software was used for image acquisition and processing.
Cell Preparation for Transplantation Experiments
For cell implantation experiments in the CNS, harvested f-MPIO-labeled NSC-Luc or NSC-Luc/eGFP were washed twice with PBS and resuspended in PBS at a concentration of 250 × 106 cells/ml. For IV cell administration, harvested NSC-Luc/eGFP and DCs were washed twice with PBS and resuspended in PBS at a concentration of 1.25 × 106 cells/ml. Cell preparations (mean viability of 80–95%) were kept on ice until transplantation experiments.
Cell Implantation in the CNS
All surgical interventions were performed under sterile conditions as previously described (8,52). Briefly, mice were anesthetized by an IP injection of a ketamine (80 mg/kg) + xylazine (16 mg/kg) mixture and placed in a mouse stereotactic frame. Cell implantation was reproducibly targeted in the right hemisphere at the following coordinates to bregma: 0 mm anterior, 2 mm lateral, and 2.5 mm ventral. For this, a midline scalp incision was made to expose the skull, and a hole was drilled in the skull using a dental drill burr at bregma at 2 mm on the right side of the midline. Thereafter, an automatic microinjector pump (kdScientific) with a 10-μl Hamilton syringe was positioned above the exposed dura. A 30-gauge needle (Hamilton) attached to the syringe was stereotactically placed through the intact dura to a depth of 2.5 mm. After 1 min of pressure equilibration, 2 μl of cell suspension was injected at a speed of 0.7 μl/min. Before needle retraction, a waiting period of 3 min was kept in order to allow for pressure equilibration and to prevent backflow of the injected cell suspension. Next, the skin was sutured, a 0.9% NaCl solution was given subcutaneously in order to prevent dehydration, and mice were placed under a heating lamp to recover. During the entire follow-up period, mice were kept in normal day-night cycle with free access to food and water.
In Vivo Bioluminescence Imaging
At different time points following intracerebral cell implantation (day 1–2, week 1 and week 2) or IV cell administration (1 min, 2 h, 6 h, and 24 h), mice were analyzed by real-time in vivo bioluminescence imaging (BLI) in order to determine the presence and/or migration of grafted NSC. For this, mice were anaesthetized by either an IP injection of a ketamine (80 mg/kg) + xylazine (16 mg/kg) mixture (for detection of intracerebral cell implants) or by a 3% isoflurane (Isoflo®) gas mixture (for biodistribution analysis), followed by an IV injection of D-luciferin (150 mg/kg body weight dissolved in PBS, Promega). Immediately after luciferin administration, mice were imaged for 3 min using an in vivo real-time ϕ-imager system (Biospace). In addition, mice that received an IV cell injection were euthanized by cervical dislocation and lungs, heart, spleen, and kidneys were removed and analyzed by ex vivo BLI. At the end of every acquisition a photographic image was obtained. The data were analyzed with Photovision software, which superimposes the BLI signal on the photographic image. The most intense BLI signal detected is shown in red, while the weakest BLI signal is shown in blue. For statistical analysis of observed BLI signals in the CNS, a region of interest (ROI) was drawn around the injection site (IS). As control, the same region was drawn on the contralateral side (CLS) of the brain. BLI results are presented as photons/s/cm2/sr, as calculated by Photovision or M3 Vision software.
In Vivo Magnetic Resonance Imaging
Imaging was performed on a horizontal magnetic resonance imaging (MRI) system (Biospec 94/20 USR, Bruker Biospin, Germany) with a magnetic field strength of 9.4 T and the standard Bruker cross coil setup being a quadrature transmit coil and a quadrature receive surface coil. Mice were anesthetized using 3% isoflurane (Isoflo®) for induction and ±1% isoflurane for maintenance in a mixture of O2:N2O (3:7) and were fixed in an animal restrainer with ear bars and a tooth bar. The surface radio frequency (RF) coil was placed on the head of the mice in a reproducible manner. Breathing rate and body temperature were continuously monitored with PCSAM software (SA Instrument rents, NY, USA). Breathing rate was maintained at 110 ± 10 breaths/min and body temperature was kept constant within a narrow range of 37 ± 0.5°C. A set of 10 coronal T2*-weighted GE images with an in-plane resolution of 66 μm2 was acquired. Sequence parameters were: repetition time (TR): 500 ms; echo time (TE): 8 ms; field of view (FOV): 17 × 17 mm2; matrix: 256 × 256; slice thickness: 0.5 mm; and slice separation 0.5 mm.
EAE Induction, Cell Administration, and Disease Scoring
EAE was induced in female C57BL/6J mice using the MOG35–55/CFA emulsion 5× PTX kit (Hooke Laboratories), according to the manufacturer's guidelines. On day 7 postinduction, mice were randomly divided into five groups: 1) EAE-induced control mice, 2) EAE-induced mice receiving a single dose of 2.5 × 105 allogeneic FVB-derived NSC-Luc/eGFP via the tail vein, 3) EAE-induced control mice receiving daily SC injections of CsA (Sandimmun®, Novartis, 10 mg/kg body weight), 4) EAE-induced mice receiving a single dose of 2.5 × 105 allogeneic FVB-derived NSC-Luc/eGFP via the tail vein followed by daily SC injections of CsA, 5) EAE-induced mice receiving a single dose of 2.5 × 105 allogeneic FVB-derived DCs via the tail vein followed by daily SC injections of CsA. Mice were blindly weighed and scored on a daily basis from day 7 to 27 postinduction. Disease score was assigned as follows: 0 = asymptomatic; 1 = partial loss of tail tonicity; 2 = complete loss of tail tonicity; 3 = partial hind limb paralysis; 4 = hind limb paralysis; 5 = breathing difficulties and/or four-leg paralysis; 6 = death in cage due to EAE. Mice scoring 5 were sacrificed in order to maintain humane end-points throughout this study. At day 8, 10, and 27 postinduction (respectively day 1, 3, and 20 following treatment), three mice of groups 1 and 4 were sacrificed for histological analysis.
Histological Analysis
Histological analysis was performed according to previously optimized procedures (8,47,52). Briefly, mice were deeply anesthetized in an induction chamber by inhalation of an isoflurane (4%), oxygen (0.5 L/min), and nitrogen (1 L/min) mixture for 2 min, and euthanized by cervical dislocation. At week 1 and 2 after intracranial implantation, whole brains were surgically removed and fixed in 4% paraformaldehyde for 2 h. Fixed brains were dehydrated in sucrose gradients (5%, 10%, 20%), frozen in liquid nitrogen, and stored at −80°C until further processing. Consecutive 10-μm-thick cryosections were cut using a micron HM5000 cryostat and observed by direct immunofluorescence microscopy in order to locate f-MPIO-labeled or eGFP-positive NSCs. Further immunofluorescence analysis was performed using the following unlabeled antibodies: a polyclonal rabbit anti-GFAP (Abcam, AB7779) in combination with Alexa Fluor® 350-labeled donkey anti-rabbit secondary antibody (Invitrogen, A10039), and a monoclonal mouse anti-mouse Tuj1 (R&D Systems, MAB1195) in combination with an Alexa Fluor® 350-labeled goat anti-mouse secondary antibody (Invitrogen, A11045). In brief, sections were rinsed with a washing buffer and incubated for 30 min in 0.1% Triton-X in PBS. Next, sections were washed and incubated for 1 h at RT in blocking serum. Subsequently, sections were incubated for 3 h at RT with the primary antibody, rinsed with washing buffer, and incubated for 1 h at RT with the secondary antibody. Next, stained sections were dehydrated and mounted. At day 1, 3, and 20 after IV cell injection in EAE-induced mice, whole brain, spinal cord, lung, heart, and spleen were removed. After fixation and dehydration, tissues were snap-frozen in liquid nitrogen and 20 representative 10-μm-thick sections were cut from each tissue and observed by direct immunofluorescence microscopy in order to locate eGFP-positive cells. Hematoxylin and eosin (H&E) and Luxol Fast Blue/Cresyl Echt violet (LFB) staining was performed on brain and spinal cord tissue to assess for lesions and demyelination, respectively. At 1 min, 2 h, 6 h, and 24 h after IV cell injection, lungs were removed and 10-μm-thick cryosections were cut after fixation and dehydration. The presence of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive apoptotic cells was investigated using the In Situ Cell Death Detection Kit (Roche, 11684817910), according to manufacturer's instructions. Inflammatory cells were detected in lung tissue using a biotin-labeled monoclonal anti-mouse CD45 antibody (eBioscience, 13–0451–85) combined with a streptavidin-horse radish-peroxidase complex (Dako, Glostrup, Denmark: 00032671) and diaminobenzidine substrate solution (DAB substrate, Dako) for visualization. Nuclei were counterstained with Carazzi's hematoxylin. All bright field and immunofluorescence analysis was performed using a standard research fluorescence microscope (Olympus BX51 fluorescence microscope) equipped with an Olympus DP71 digital camera. Olympus Cell-F Software was used for image acquisition and processing.
Statistical Analysis
For in vitro cell differentiation studies and in vivo BLI analysis, results are expressed as mean ± SE and comparisons were validated using Student's t -test with a value of p < 0.05 considered to be statistically significant. For EAE clinical score data, results are presented by box plots and day-per-day comparisons between the different groups were validated by the Mann-Whitney U-test (in SPPS) with a value of p < 0.0002 (for 0.05 critical significance level according to Bonferroni correction) considered to be statistically significant. The individual effects of CsA, NSCs, and DCs in function of time were validated by the Generalized Estimating Equations (GEE) model (in SPSS) with a value of p < 0.0125 (for 0.05 critical significance level according to Bonferroni correction) considered to be statistically significant.
Results
Isolation and Culture of NSC From ROSA26-L-S-L-Luciferase Mice
In the first part of this study, we derived adherently growing NSC cultures starting from embryonic brains of ROSA26-L-S-L-Luciferase transgenic mice (Fig. 1A). In a first step, neurosphere cultures were initiated from single cell suspensions of embryonic brains (Fig. 1B, left picture). Next, adherently growing NSC cultures were allowed to grow on fibronectin-coated culture flasks (Fig. 1B, right picture). At passage 2, when cultures became homogenous for cells with NSC morphology, cultures were screened for presence and/or absence of specific membrane proteins characteristic for different (stem) cell types. Flow cytometric analysis indicated that the cultured populations displayed uniform expression of neural antigens A2B5 and NCAM, without detectable expression of hematopoietic (CD45, c-kit), mesenchymal (Sca-1), and endothelial (CD31) membrane proteins (data not shown). Phenotypical properties of these adherent cultures of NSCs remained unchanged for at least 38 passages (data not shown).

Isolation and characterization of luciferase-expressing NSCs from ROSA26-L-S-L-Luciferase mice. (A) Molecular organization of the ROSA26 locus in ROSA26-L-S-L-Luciferase transgenic mice. (B) Representative pictures of cultured NSCs taken under phase contrast microscopy. Left: neurosphere culture of NSCs. Right: adherent culture of NSCs. Scale bars: 200 μm. (C) Adherent NSC cultures were nonlipofected (NO LIPO, upper dot plot) or lipofected with eGFP mRNA (LIPO eGFP, lower dot plot), and were analyzed by flow cytometry for eGFP fluorescence (x-axis) versus viability (GelRed-staining, y-axis) after 24 h of culture. The percentage indicated in the lower left quadrant is the number of viable eGFP-negative cells. The percentage indicated in the lower right quadrant is the number of viable eGFP-positive cells. The percentages indicated in the upper left and upper right quadrant are numbers of nonviable cells. Representative dot plots are shown for six independent experiments. (D) In vitro luminescence assay on parental NSCs, on Cre-recombined luciferase-expressing NSCs (NSC-Luc), and on eGFP-encoding lentiviral vector-transduced NSC-Luc (NSC-Luc/eGFP). Results are presented as mean luminescence signal ± SE of four independent measurements. (E) Representative flow cytometric analysis showing expression pattern of membrane proteins on NSC-Luc (i.e., expression of A2B5 and NCAM, but no expression of Sca-1 and CD45). Open histograms: control. Filled histograms: specific antibody staining. Representative histograms are shown for 10 independent phenotyping experiments at different passages.
Activation of Luciferase Expression in NSCs Derived From ROSA26-L-S-L-Luciferase Mice
Next, in order to activate expression of the luciferase reporter gene in NSCs derived from ROSA26-L-S-L-Luciferase transgenic mice, the “floxed” neomycin resistance cassette was excised by Cre recombinase. First, we optimized a transfection protocol using Lipofectamine 2000 in order to introduce mRNA into cultured NSC. Flow cytometric analysis of NSCs lipofected with mRNA encoding the eGFP indicated efficient transgene expression in up to 60% of viable cells 24 h postlipofection (Fig. 1C). Next, NSCs were lipofected with mRNA encoding the Cre recombinase protein in order to activate luciferase expression. A polyclonal luciferase-expressing NSC culture was obtained (further designated as NSC-Luc), which demonstrated stable luciferase activity for at least 27 passages, as shown by a standard in vitro luminescence assay (Fig. 1D). In addition, flow cytometric analysis indicated that cultured NSC-Luc displayed the same expression pattern of membrane proteins compared to nonrecombined parental NSC cultures (Fig. 1E).
Characterization of NSC-Luc Derived From ROSA26-L-S-L-Luciferase Mice
Next, in order to demonstrate that cultured NSC-Luc have the capacity to differentiate into both glial (GFAP+ astrocytes) and neuronal (Tuj1+ neurons) cell lineages, in vitro differentiation studies were performed. For immunofluorescence analysis (Fig. 2), we compared the expression pattern of SOX2, BLBP, GFAP, and Tuj1 on undifferentiated NSC-Luc, in vitro differentiated NSC-Luc, BMSCs (negative control), primary neuron cultures (positive control), and primary astrocyte cultures (positive control). While undifferentiated NSC-Luc were immunoreactive for SOX2, BLBP, GFAP, but not for Tuj1, in vitro differentiated NSC-Luc cultures clearly showed the morphological presence of astrocytes and neurons. Astrocytes differentiated from NSC-Luc were immunoreactive for SOX2, BLBP, and GFAP, comparable to the expression demonstrated in primary astrocyte cultures. Although marker expression between astrocytes and NSC-Luc was similar, differentiated astrocytes from NSC-Luc underwent clear morphological changes compared to undifferentiated NSC-Luc. Neurons differentiated from NSC-Luc were immunoreactive for Tuj1, but not GFAP and BLBP, comparable to the expression demonstrated in primary neuron cultures. SOX2 expression in neurons differentiated from NSC-Luc was lower compared to the expression in astrocytes differentiated from NSC-Luc, while it was absent in primary neuron cultures. The low expression of SOX2 in neurons differentiated from NSC-Luc might be due to incomplete neuronal differentiation. Quantification of the number of astrocytes and neurons in differentiated NSC-Luc cultures indicated 81 ± 4% of astrocytes and 18 ± 4% of neurons.

In vitro differentiation potential of NSC-Luc. Row 1: Bright field pictures of undifferentiated luciferase-expressing NSCs (NSC-Luc column), in vitro differentiated luciferase-expressing NSC (NSC-Luc-D column), bone marrow-derived stromal cells (BMSC column, negative control for immunofluorescence staining), primary neuron cultures (NEURON column, positive control for immunofluorescence staining), and primary astrocyte cultures (ASTROCYTE column, positive control for immunofluorescence staining). Row 2: Immunofluorescence staining for SOX2 (red) and Tuj1 (green). Row 3: Immunofluorescence staining for BLBP (red), Tuj1 (green), and nucleus (DAPI, blue). Row 4: Immunofluorescence staining for GFAP (red), Tuj1 (green), and nucleus (DAPI, blue). Representative pictures were chosen from 20 independent in vitro differentiation experiments. Scale bars: 50 and 100 μm, as indicated.
Fluorescent Labeling of NSC-Luc Cultures to Allow Cell Graft Detection by Histology
Following characterization of our NSC-Luc cultures, initial transplantation experiments of these cultures into the CNS of syngeneic immune-competent mice indicated their survival capacity in vivo, as monitored by real-time in vivo BLI (data not shown). However, subsequent histological analysis could not unambiguously confirm the presence of grafted cells due to the low specificity of existing antiluciferase antibodies (data not shown). Therefore, before initiating new cell transplantation experiments, two alternative strategies to fluorescently label NSC-Luc were investigated in vitro. First, in order to be able to localize NSC-Luc cell grafts in vivo by histology, NSC-Luc cultures were loaded with 0.96 μm f-MPIO containing the Dragon Green fluorophore. Flow cytometric analysis of f-MPIO-labeled NSC-Luc indicated a labeling efficiency of >90% without significant cell mortality (FACS analysis) (Fig. 3A). In addition, immunofluorescence staining of f-MPIO-labeled NSC-Luc with NCAM suggested the intracellular localization of the f-MPIO particles (immunofluorescence picture) (Fig. 3A). Alternatively, NSC-Luc were transduced using a lentiviral vector encoding the eGFP and the Pac resistance gene. Following puromycin selection, a polyclonal Luciferase-, eGFP-, and Pac-expressing NSC line was obtained (further named as NSC-Luc/eGFP; immunofluorescence picture) (Fig. 3B). Flow cytometric analysis of NSC-Luc/eGFP demonstrated eGFP transgene expression in >95% of viable cells (FACS analysis) (Fig. 3B). Moreover, phenotypical properties were not influenced following lentiviral-transduction of NSC-Luc (data not shown).

Fluorescent labeling of NSC-Luc from ROSA26-L-S-L-Luciferase mice. (A) Immunofluorescence picture: Representative immunofluorescence analysis showing NCAM-expressing NSC-Luc (blue) labeled with fluorescent f-MPIO particles (green). A representative picture was chosen from three independent experiments. Inset dot plot: NSC-Luc labeled with f-MPIO and analyzed by flow cytometry for Dragon Green fluorescence (x-axis) versus viability (GelRed staining, y-axis) after 48 h of culture. A representative dot plot is shown for five independent experiments. Scale bar: 100 μm. (B) Immunofluorescence picture: Direct eGFP fluorescence of eGFP-encoding lentiviral vector-transduced NSC-Luc. A representative picture was chosen from three independent experiments. Inset dot plot: NSC-Luc/eGFP cells were analyzed by flow cytometry for eGFP fluorescence (x-axis) versus viability (GelRed staining, y-axis) at several passages. A representative dot plot is shown for six independent measurements. Scale bar: 200 μm.
BLI, MRI, and Histological Analysis of f-MPIO-Labeled NSC-Luc and NSC-Luc/eGFP Implants in the CNS of Syngeneic Immune-Competent Mice
Next, in order to investigate whether cultured f-MPIO-labeled NSC-Luc and NSC-Luc/eGFP survive in vivo upon intracerebral implantation, 5 × 105 cells were grafted in the CNS of syngeneic immune competent ROSA26-L-S-L-Luciferase transgenic mice. First, survival of cell implants was monitored by real-time in vivo BLI at several time points postimplantation (Fig. 4A). For 25 mice implanted with either f-MPIO-labeled NSC-Luc (n = 18) or NSC-Luc/eGFP (n = 7) grafts in the CNS, a clear BLI signal (i.e., signal intensity at least doubled when comparing the IS with the CLS) was detected in 19/20 mice analyzed at day 1–2 postimplantation (p = 0.012), in 9/10 mice analyzed at week 1 postimplantation (p = 0.017), and in 19/19 mice analyzed at week 2 postimplantation (p = 0.017). Quantitative analysis of the obtained BLI signals clearly demonstrated that in vivo survival of f-MPIO-labeled NSC-Luc and NSC-Luc/eGFP can be successfully monitored with BLI until week 2 postimplantation (pooled data from both groups) (Fig. 4B). Next, because BLI lacks spatial resolution, we additionally investigated whether f-MPIO-labeled NSC-Luc grafts can also be localized by MRI. Using MRI, we were able to detect f-MPIO particles, appearing as a dark spot on T2*-weighted MRI images, in the CNS at week 2 posttransplantation in 3/3 mice analyzed (Fig. 4C). These data demonstrate that MRI and BLI can be combined on the same animals in order to noninvasively provide data regarding localization and viability of f-MPIO-labeled NSC-Luc implants. Furthermore, histological analysis was performed at week 2 postimplantation. Randomly chosen animals were sacrificed and dissected brains were examined for cell graft localization and differentiation. The presence of f-MPIO-labeled NSC-Luc and NSC-Luc/eGFP grafts could clearly be demonstrated by direct immunofluorescence on unstained slides (f-MPIO and eGFP; Fig. 4D, E). Next, grafted cells were analyzed for in vivo differentiation potential. Combined immunofluorescence staining (blue) and f-MPIO or eGFP fluorescence (green) (MERGE; Fig. 4D, E) clearly indicates in vivo differentiation of NSC into astrocytes (GFAP; Fig. 4D, E), but not into neurons (Tuj1; Fig. 4D, E).

BLI, MRI, and histological analysis of f-MPIO-labeled and eGFP-expressing NSC-Luc following implantation in the CNS of syngeneic immune-competent mice. (A) In vivo BLI of grafted f-MPIO-labeled NSC-Luc. A representative time course image is shown at week 1 postimplantation and week 2 postimplantation. The most intense BLI signal detected is shown in red, while the weakest BLI signal is shown in blue. The intensity values are given in photons/s. (B) Statistical analysis of observed BLI signals at different time points postimplantation (day 1–2, week 1, and week 2). The BLI intensity values are indicated as mean number of photons/s/cm2/sr ± SE. (C) In vivo MRI of f-MPIO-labeled NSC-Luc. Implanted NSC-Luc appear as a dark spot (indicated by white arrow) on consecutive MRI images at different distances from bregma. Scale bars: 5 mm. (D, E) Histological analysis of f-MPIO-labeled NSC-Luc and NSC-Luc/eGFP at week 2 postimplantation. Unstained tissue sections (f-MPIO and eGFP) clearly show the localization and general appearance of the implantation site. Tuj1-stained slides and merged pictures indicate the absence of in vivo neuronal differentiation of NSC-Luc grafts. GFAP-stained slides and merged pictures indicate the in vivo astrocyte differentiation of NSC-Luc grafts. Representative pictures were chosen from four cell-implanted brains analyzed for each condition. Scale bars: 500 μm.
Therapeutic Potential of Allogeneic NSC Transplantation in EAE Mice
In the second part of this study we investigated whether IV administration of allogeneic adherently cultured NSC has a positive effect on disease outcome in EAE-induced mice. For this, EAE was induced in female C57BL/6J mice and mice were randomly divided into five groups on day 7 postinduction: 1) EAE-induced control mice (EAE, n = 62), 2) EAE-induced mice receiving a single dose of 2.5 × 105 allogeneic FVB-derived NSC-Luc/eGFP via the tail vein (EAE + NSC, n = 15), 3) EAE-induced control mice receiving daily SC injections of CsA (EAE + CsA, n = 9), 4) EAE-induced mice receiving a single dose of 2.5 × 105 allogeneic FVB-derived NSC-Luc/eGFP via the tail vein followed by daily SC injections of CsA (EAE + CsA + NSC, n = 42), 5) EAE-induced mice receiving a single dose of 2.5 × 105 allogeneic FVB-derived DCs (negative control cell population) via the tail vein followed by daily SC injections of CsA (EAE + CsA + DC, n = 25). Mice were then evaluated on a daily basis until week 3 posttreatment (Fig. 5A). Following day-to-day comparison of the different groups using the MWU statistical test, no statistical difference in disease course was observed between the different treatment groups. Of note, mice receiving allogeneic NSCs without CsA treatment displayed a significant earlier onset of disease. However, this might be explained by partial loss in tail tonicity due to ongoing allogeneic immune responses against remaining cells in the tail vain, which resembles scoring level 1 on the 5-point EAE scoring scale (Fig. 5A). Alternatively, using the GEE statistical method for comparing the disease course over time, a clear effect could be attributed to CsA treatment (p = 0.001), but not to NSC or DC administration (p = 0.063 andp = 0.521, respectively). In addition, at day 1, 3, and 20 following NSC-Luc/eGFP administration, three mice of the EAE + CsA + NSC group were sacrificed in order to examine spinal cord, brain, lung, heart, and spleen for the presence of eGFP-expressing cells. While spinal cord lesions could clearly be demonstrated on H&E- and LFB-stained sections, no eGFP-expressing NSC-Luc/eGFP could be detected by direct immunofluorescence analysis of following sections (week 3 posttreatment) (Fig. 5B). Furthermore, in none of the other tissues or at other time points analyzed were we able to detect eGFP-expressing NSC-Luc/eGFP (or progeny) by direct immunofluorescence analysis of tissue sections (data not shown).
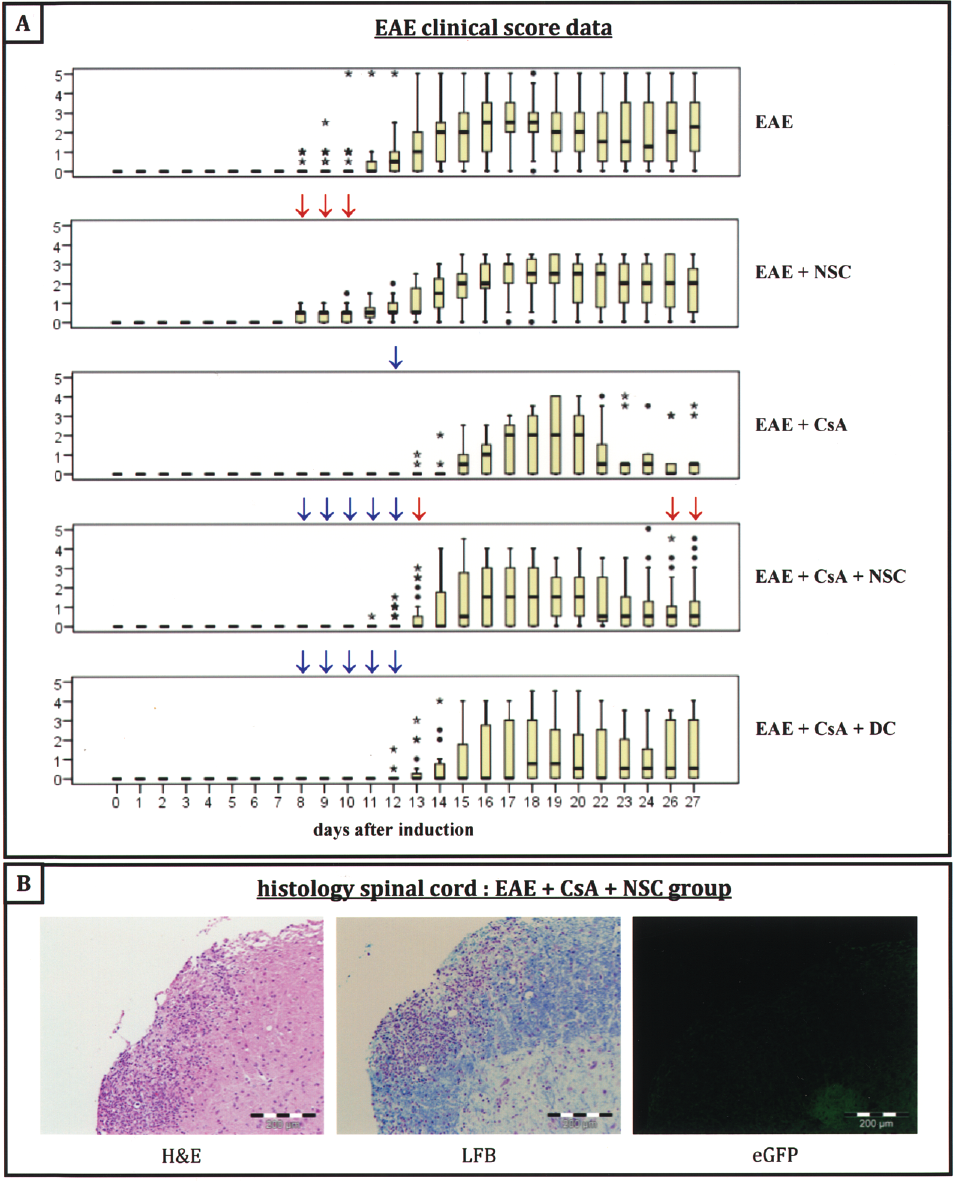
Therapeutic potential of IV administered allogeneic NSCs in EAE mice. (A) Box plots indicating clinical scores of EAE-induced female C57BL/6J mice (EAE, first box plot), EAE-induced female C57BL/6J mice treated with allogeneic FVB mouse-derived neural stem cells at day 7 postinduction (EAE + NSC, second box plot), EAE-induced female C57BL/6J mice treated with daily injections of CsA at day 7 postinduction (EAE + CsA, third box plot), EAE-induced female C57BL/6J mice treated with allogeneic FVB mouse-derived neural stem cells and daily injections of CsA at day 7 postinduction (EAE + CsA + NSC, fourth box plot), EAE-induced female C57BL/6J mice treated with allogeneic FVB mouse-derived dendritic cells and daily injections of CsA at day 7 postinduction (EAE + CsA + DC, fifth box plot). Extreme values are indicated by an asterisk (*) and outliers are indicated by a circle (•). For each treatment group, a day-to-day statistical significance of p < 0.0002 versus the control EAE group, as determined by the MWU test, is indicated by small red arrows (↓) above each box plot. For each treatment group, a day-to-day statistical significance of p < 0.0002 versus the EAE + NSC group, as determined by the MWU test, is indicated by small blue arrows (↓) above each box plot. (B) Histological analysis of lesions in the spinal cord at day 20 after EAE induction. Spinal cord sections were first stained with H&E (left picture) and LFB (middle picture) to localize demyelination. No eGFP+ cells could be detected in sections adjacent to the lesions (right picture). Scale bars: 200 μm.
Intravenously Administered NSCs Accumulate in the Lung and Cause Inflammation and Apoptosis in Lung Tissue
In order to investigate the absence of migrated NSC-Luc/eGFP at sites of neuroinflammation (spinal cord, brain) at every time point analyzed by histological analysis (day 1, 3, and 20 postinjection), we examined the biodistribution of NSC-Luc/eGFP upon IV administration using the above-described and optimized in vivo BLI procedure. For this, following IV administration of 2.5 × 105 FVB-derived NSC-Luc/eGFP in C57BL/6J mice, in vivo BLI biodistribution analysis was performed after 1 min, 2 h, 6 h, and 24 h (Fig. 6A). The data clearly demonstrate an accumulation of viable NSC-Luc/eGFP in the lung at 1 min, 2 h, and 6 h following IV injection, while no signals could be detected by 24 h postinjection. Moreover, these results were confirmed by ex vivo BLI biodistribution analysis on lung, heart, liver, spleen, and kidney (Fig. 6B). These data demonstrate that IV administered NSC-Luc/eGFP accumulate in the lung and most likely die within 24 h. Next, we aimed to further investigate the effect of cell accumulation in the lung. For this, histological analysis was performed on lungs dissected from control mice and mice 6 h following IV administration of NSC-Luc/eGFP or DCs (Fig. 6C). In contrast to control lungs, large numbers of CD45+ inflammatory cells and TUNEL+ apoptotic cells were observed in lungs dissected from NSC-Luc/eGFP administered mice. In contrast, the latter was not observed upon IV administration of suspension cultured DCs. Finally, lung tissue of EAE mice that received IV administered NSC-Luc/eGFP was analyzed for the presence of inflammatory and apoptotic cells. Results indicate the presence of large numbers of CD45+ inflammatory cells and TUNEL+ apoptotic cells (Fig. 6D) at day 1 postinjection, indicating that lack of regenerative effect following IV administration of NSC-Luc/eGFP is most likely due to cell accumulation and death in the lung, followed by inflammation and apoptosis in lung tissue.

Intravenously administered NSC accumulate in the lung and provoke inflammation and apoptosis in lung tissue. (A) In vivo BLI of IV administered NSC-Luc/eGFP. A representative image is shown at 1 min, 2 h, 6 h, and 24 h postinjection. Mice were imaged for 1 min using an in vivo real-time (p-imager system. The intensity values are given in photons/s. (B) Ex vivo BLI of IV administered NSC-Luc/eGFP. Following in vivo BLI, mice were sacrificed and lung, heart, spleen, liver, and kidneys were removed and analyzed for 1 min. A representative image of the organs is shown at 6 h and 24 h postinjection. (C) Histological analysis of control lung tissue (left panel), and lung tissue of mice that received an IV injection of NSC-Luc/eGFP (middle panel) or DCs (right panel) at 6 h postimplantation. A large number of apoptotic cells is seen in lung tissue of mice that received an IV injection with NSC-Luc/eGFP (middle panel, TUNEL). No apoptotic cells could be detected in lung tissue of control mice (left panel, TUNEL) and mice that received an IV injection of DCs (right panel, TUNEL). In addition, large numbers of CD45+ inflammatory cells were observed in lung tissue dissected from NSC-Luc/eGFP-injected mice (middle panel, CD45). In contrast, no inflammatory cells could be observed in lung tissue of control mice (left panel, CD45) and mice that received an IV injection of DCs (right panel, CD45). Scale bars: 100 μm. (D) Histological analysis of lung tissue of EAE mice that received an IV injection of NSC-Luc/eGFP. Results also indicate the presence of large numbers of TUNEL+ apoptotic cells (left picture) and CD45+ inflammatory cells (right picture) in lung tissue at 24 h postinjection. Scale bars: 100 μm.
Discussion
In the first part of this study, we derived adherently growing NSC cultures from embryonic brains of ROSA26-L-S-L-Luciferase transgenic mice. In contrast to traditional neurosphere cultures of NSCs, the adherent NSC cultures obtained in this study displayed a uniform morphology and expression of A2B5, NCAM, BLBP, GFAP, and SOX2 markers (Figs. 1B, E and 2). These phenotypical properties are similar to several literature reports suggesting that NSCs are closely related to the radial glial lineage and have multiple characteristics of astrocytes (1,14,25,33). Moreover, in vitro differentiation studies were performed demonstrating both astrocyte and neuron differentiation of our cultured adherently growing NSCs, further supporting their stem cell identity (Fig. 2). In addition, recent studies also demonstrated the differentiation capacity of adherently cultured NSCs into oligodendrocytes upon specific growth factor stimulation (19,51).
Next, in order to demonstrate the in vivo grafting capacity of our cultured NSCs by BLI, luciferase expression was activated following transfection of cultured NSCs with mRNA encoding the Cre recombinase protein (Fig. 1C, D). While initial transplantation studies demonstrated in vivo survival of our cultured NSC-Luc cells as demonstrated by BLI, we were unable to confirm these results by histological analysis due to inconclusive staining with an antiluciferase antibody (data not shown). We therefore loaded our cultured NSC-Luc with 0.96 μm f-MPIO (Fig. 3A). The advantage of these particles over nonfluorescent MPIO is their relatively easy detection by flow cytometry in order to determine loading efficiency and by histology in order to determine cell implant localization. Moreover, labeling of NSC-Luc with f-MPIO particles did not interfere with in vivo cell survival upon implantation in the CNS of immunocompetent mice (Fig. 4A, B). In addition, MRI can detect these f-MPIO upon implantation of f-MPIO-labeled NSC-Luc in the CNS of mice (Fig. 4C). However, for the latter, further technical development of such particles will be needed in order to obtain optimal MRI images. Current MRI images result in an approximately 10 times overestimation of implant size due to MPIO susceptibility artifacts. The latter is most likely due to the high iron content (0.4 pg/particle) of these particles. Therefore, most likely one needs to develop novel particles, which display the same fluorescence intensity, but contain less iron, in order to obtain correlation between MRI and histology (34).
Nevertheless, the currently used fluorescent MPIO particles are very suitable for cell graft detection by histology. Characterization of f-MPIO-labeled NSC-Luc grafts revealed an in vivo differentiation potential mainly into GFAP+ astrocytes, but not into Tuj1+ neurons (Fig. 4D). Currently, we do not know whether the limited in vivo differentiation potential, compared to the in vitro differentiation potential (Fig. 2), is intrinsic to our cultured (f-MPIO-labeled) NSC-Luc. Recent studies demonstrated the microenvironment can determine the differentiation fate of engrafted NSCs (35). Depending on the time of transplantation (acute or delayed) in an experimental spinal cord injury model, NSCs integrate in and participate to the formation of a glial scar, or integrate around the scar and differentiate into migrating neurons. It has also been reported that NSCs transplanted into the uninjured brain undergo targeted migration after stroke onset, and remain latent in an undifferentiated state upon implantation (23). We cannot exclude that some of the implanted NSC-Luc did not undergo differentiation, as some of the currently validated markers (SOX2, BLBP, GFAP) used in this study are shared between NSCs and astrocytes (Fig. 2). Further studies will have to address this topic.
One interesting observation with the above-described histology results is that both GFAP+ f-MPIO+ and GFAP+ f-MPIO- cells can be observed (Fig. 4D). This can be explained by the fact that cell division might have occurred and/or labeled cells have lost their f-MPIO in vivo. Alternatively, one might be looking at both transplanted (astrocyte-differentiated) f-MPIO-labeled NSC-Luc (GFAP+ f-MPIO+) and endogenous astrocytes (GFAP+ f-MPIO-). However, due to the absence of a cell viability marker for histological analysis (e.g., eGFP), we were unable to make final conclusions regarding this observation. In this context, we have recently shown that both luciferase and eGFP can be expressed by syngeneic BMSC grafts in the CNS of immunocompetent mice without inducing immune-mediated rejection (8). Therefore, NSC-Luc were transduced using the same lentiviral vector in order to obtain eGFP-expressing NSC-Luc/eGFP (Fig. 3B). Again, in vivo survival of NSC-Luc/eGFP following implantation in the CNS of immune-competent mice was demonstrated by BLI (Fig. 4B) and histological analysis confirmed the limited in vivo differentiation into GFAP+ astrocytes, but not into Tuj1+ neurons (Fig. 4D). Moreover, we here can confirm that grafted NSC-Luc/eGFP (GFAP+ eGFP+) become (slightly) encapsulated by endogenous astrocytic scar tissue (GFAP+ eGFP-). Currently, we do not yet know how the development of this scar tissue around implanted NSC will influence long-term in vivo graft survival and/or cellular integration.
Summarizing the first part of this study, we have developed several experimental procedures in order: i) to culture, characterize, and fluorescently label adherently growing uniform NSC cultures from embryonic brain of mice; ii) to noninvasively demonstrate NSC graft survival and location by respectively BLI and MRI; and iii) to demonstrate by histological analysis (limited) in vivo differentiation potential of NSCs and potential endogenous astrocyte activation upon cell implantation.
Having optimized the above-described cell culture and imaging methodologies, we aimed to further investigate whether adherently cultured NSC have a potential capacity to be used in regenerative medicine. Following establishment of a reproducible EAE model, four potential treatment options were investigated (Fig. 5A). Using the GEE statistical method for comparing the disease course over time, a clear effect could be attributed to CsA treatment, but not to NSC or DC administration. Next, in order to understand these results, histological analysis indicated the absence of NSC-Luc/eGFP cells at sites of neuroinflammation at day 1, 3, and 20 posttransplantation (Fig. 5B). This suggested that the lack of therapeutic benefit most likely can be attributed to the inability of IV administered NSC-Luc/eGFP to home to sites of injury in the CNS.
In order to further document the latter, in vivo biodistribution analysis of IV-administered NSC-Luc/eGFP was performed using in vivo and ex vivo BLI. Here we demonstrate that IV-administered NSC-Luc/eGFP are retained in lung capillaries from 1 min postinjection on (Fig. 6A). While at 6 h postinjection few surviving cells can be detected in the lung, no BLI signal could be detected 24 h postinjection (Fig. 6A, B). The latter explains why no NSC-Luc/eGFP could be detected by histological analysis at sites of neuroinflammation (Fig. 5B). Several other reports previously indicated the retention of BMSCs in lung capillaries upon IV administration (17,29,30). In our hands, IV administration of BMSCs in mice leads to death within 1 min postinjection due to asphyxia (data not shown). Because NSCs are much smaller cells compared to BMSCs, we did not expect to observe retention in lung capillaries. While in the presented data we used adherently cultured NSCs, similar results were observed following IV administration of dissociated neurosphere cultures (data not shown). In addition, cell retention in the lung was also observed following IV and intracoronary (IC) administration of autologous NSC-Luc/eGFP in syngeneic mice (data not shown). Moreover, retention of NSC-Luc/eGFP in the lung leads to massive inflammation and apoptosis in lung tissue (Fig. 6C, D). These data should warrant caution for further studies and/or clinical trials aiming to develop NSC- or BMSC-based therapies when cells are IV administered. Finally, in order to investigate why NSCs and BMSCs are retained in the lung following IV administration, we examined lung tissue of mice following IV administration of bone marrow-derived DCs (Fig. 6C), which are expanded in suspension culture. In this condition, although we could not track the cells due to the absence of Luc/eGFP expression, no inflammatory and/or apoptotic cells were observed in lung tissue. This suggests that cell retention in the lung most likely is a negative side effect occurring with in vitro cultured cells expanded on substrate (e.g., adherently cultured BMSCs and NSCs) or in spherical culture (e.g., neurospheres), but not with suspension cultured cells (e.g., DCs).
Summarizing the second part of this study, we have investigated whether IV administration of allogeneic NSCs under CsA treatment has a potential benefit for treatment of EAE. i) Although CsA clearly improves EAE clinical score, no additional benefit could be attributed to NSC administration. ii) We observed cell retention in lung capillaries directly postinjection, resulting in inflammation and apoptosis in lung tissue. The latter should be taken into account during further development of cellular therapies.
Footnotes
Acknowledgments
We acknowledge helpful assistance from August Van Laer (Laboratory of Experimental Surgery) with animal handling and surgical procedures and from Frank Rylant and Ingrid Bernaert (Laboratory of Pathology) with histological techniques. We thank Geoffrey De Visscher from the Bio-Imaging Lab & Biomedical Microscopic Imaging Core Facility for help with statistical analysis. Lentiviral vectors were produced by the Viral Vector Core, Division of Molecular Medicine, K.U. Leuven. This work was supported by research grant G.0132.07 (granted to Z.B., P.J., D.Y., and E.V.M.) and 1.5.021.09.N.00 (granted to P.P.) of the Fund for Scientific Research–Flanders (FWO-Vlaanderen, Belgium), by research grants BOF-KP 2006 (granted to S.C.) and BOF-NOI 2006 (granted to P.P. and S.C.) from the University of Antwerp, by research grant BRAINSTIM of the Flemish Institute for Science and Technology (granted to Z.B., V.B., and A.V.D.L.), in part by a Methusalem research grant from the Flemish government (granted to H.G. and Z.B.), in part by EC-FP6-NoE DiMI and LSHB-CT-2005-512146 NoE (granted to V.B. and A.V.D.L.), in part by LSHC-CT-2004-503569 EMIL (granted to A.V.D.L.), and by the Fund for Cell Therapy of the Antwerp University Hospital. Nathalie De Vocht holds a Ph.D. studentship from the FWO-Vlaanderen and Peter Ponsaerts is a postdoctoral fellow of the FWO-Vlaanderen.