
Abstract
Reliable noninvasive methods are needed to monitor cell engraftment and graft survival after hepatocyte transplantation. Superparamagnetic iron oxide nanoparticles (SPIOs) have been shown to accumulate in various types of cells, and are currently the labeling agent of choice for cellular magnetic resonance imaging (MRI). However, for successful clinical translation to hepatocyte transplantation, it is important that hepatocytes maintain their viability and synthetic function after labeling. In this study, primary human hepatocytes were incubated with increasing concentrations of clinical grade SPIOs for different time intervals. SPIOs uptake was confirmed by light and fluorescence microscopy, and intracellular iron content quantified by a colorimetric ferrozine-based assay. Studies were performed to determine if labeling affected cell viability and function. Intracellular iron concentrations increased in a time- and dose-dependent manner after incubation with SPIOs. Labeling had minimal short-term effects on cell attachment and mitochondrial function. However, exposure of hepatocytes to SPIOs resulted in a dose- and time-dependent reduction in protein synthesis. Cell labeling for 16 h had no significant effect on hepatocyte-specific function, but longer periods of incubation resulted in a dose-dependent decrease in albumin production. Hepatocytes incorporated SPIOs at sufficient levels for in vitro detection on a 7-T MRI imaging system, with a minimum of 2,000 SPIO-labeled cells/μl detected by a decreased T2 relaxivity compared to controls. Intrasplenic transplantation of human hepatocytes labeled with 50 μg Fe/ml of SPIOs was performed in nonobese diabetic/severe combined immune deficiency (NOD-Scid) mice. Recipient livers showed a clear decrease in signal intensity on T2*-weighted MR images when compared to controls, allowing detection of hepatocytes. With further experiments to optimize the conditions for labeling human hepatocytes, it should be possible to apply this technique to track hepatocyte transplantation in patients with liver disease.
Keywords
Introduction
With recent advances in the field of cell therapy, there has been an increasing interest in the use of magnetic resonance imaging (MRI) to track and monitor transplanted cells and cellular processes in vivo (27). Hepatocyte transplantation (HT) is one of the cell therapies that would benefit from this technique (32). Transplantation of hepatocytes has been studied as a therapeutic alternative or as a bridge to orthotopic liver transplantation (9,12), and although clinical trials in patients with acute liver failure (ALF) and liver-based metabolic disorders have proven HT safe, no complete and sustained correction of liver disease by this technique has yet been achieved. This limited clinical efficacy might be the result of substantial hepatocyte loss after transplantation. Potential causes for this initial cell loss are apoptosis (13), tissue factor-dependent activation of the coagulation cascade (39), and/or acute humoral rejection of transplanted hepatocytes (17). The early phases of HT are critical and initial monitoring of graft function is usually indirect, determined by the patient's metabolic improvement, or dependent on analysis of multiple invasive liver biopsies (23,38,40). However, metabolic changes may take a few weeks to become evident and can be difficult to assess in patients with ALF due to frequent concurrent multiorgan failure. To date, scintigraphic imaging using indium-111-labeled hepatocytes has been the only method reported for short-term noninvasive monitoring of clinical HT (4). This represents a major limitation for the development of clinical HT.
MRI offers high anatomical detail, combined with the possibility of serial imaging, but requires an efficient labeling method with suitable contrast agents before transplantation to allow tracking of grafted cells within the host tissue (27). Although gadolinium-based agents potentially provide an unequivocal positive contrast, they have been found to increase reactive oxygen species (ROS) in neural stem cells and to impair the repair process in stroke-lesioned brains (6,26). In contrast, superparamagnetic iron oxide nanoparticles (SPIOs) are the most common class of contrast agents used for cellular labeling and imaging, and various types of cells have been efficiently labeled and tracked in vivo (8,37). It is generally accepted that iron oxide particles are well tolerated in vivo. After IV administration, most SPIOs are rapidly cleared from the blood by reticuloendothelial system (RES) cells of the liver, spleen, lymph nodes, and bone marrow (41), appearing as regions of low signal intensity (hypointense) on T2*- and T2-weighted MRI sequences, creating a negative contrast. Once internalized by cells, SPIOs are compartmentalized within the lysosomes, metabolized, and recycled in the normal iron pool (7).
In order to be detected by MRI, cells of interest must be labeled with relatively large amounts of contrast agent. In addition, slowly dividing cells and those that lack substantial phagocytic activity require incubation with high concentrations of SPIOs for extended periods of time for an efficient labeling. While it has been reported in several studies that SPIO labeling does not appear to affect viability or other cellular properties (1,15,28), some recent reports have shown that iron oxide particles can have a dramatic effect upon cell behavior and morphology (3,21,29,31). As a result, there is a need for detailed studies on the biological effects of SPIO labeling, which may vary according to different cell types.
Here we examined and quantified the specific effects that clinically approved SPIOs (Endorem®) have on primary human hepatocytes in short- and long-term cultures, evaluate whether labeled cells retain their ability to perform normal cellular functions, and if they can be detected by MRI.
Materials and Methods
Human Hepatocyte Isolation and Culture
Hepatocytes were isolated from 13 liver resection tissues (median donor age 65 years; range 55–73 years) and 13 unused donor liver segments/lobes (42 years; 13–59 years). All tissues were consented for research in accordance with the Research Ethics Committee of King's College Hospital. Cell isolation was carried out using a modified two-step collagenase perfusion technique (24) and hepatocytes were purified by low speed centrifugation at 50 × g for 5 min at 4°C. The isolated cell fraction used for in vitro studies was >90% hepatocytes by morphological and immunocytochemical analysis. Briefly, cytospins were fixed in acetone and endogenous peroxidase blocked with 0.3% H2O2 in methanol for 15 min. Cells were blocked and permeabilized with 5% fetal calf serum (FCS) in Tris-buffered saline containing 0.1% Triton X-100 (Sigma-Aldrich, UK) for 30 min, followed by incubation with goat anti-human antibody against albumin horseradish peroxidase-conjugated (dilution 1:200; Bethyl Laboratories, USA) and color development using diaminobenzidine (KPL, USA). HeLa cells were used as negative controls. The mean cell yield of isolated hepatocytes was 4.4 ± 0.74 × 106 cells per gram of liver tissue with 71 ± 2% viability, as determined by the trypan blue (0.4%) exclusion method. Collagen-coated tissue culture plates were seeded with hepatocytes (16 × 104 viable cells/cm2) and cultured at 37°C in 95% O2/5% CO2. Standard hepatocyte culture medium consisted of Williams' E medium (WEM), supplemented with 10 mM HEPES (Cambrex, UK), 10% heat-inactivated FCS, 2 mM L-glutamine (Invitrogen, UK), 0.1 μM dexamethasone, 0.1 μM insulin, penicillin (50 IU/ml), and streptomycin (50 μg/ml) (Sigma-Aldrich, UK). Plating efficiency was determined by measuring protein content (22) of attached cells and that of the initial number of viable cells seeded, and the mean value was 54 ± 4%.
In Vitro Hepatocyte Labeling
SPIO nanoparticles, Endorem® (Guebert, France), a clinically approved liver MRI contrast agent, was used to label cells. This contrast agent consists of dextran-coated iron oxide particles (80–150 nm in size) and has a total iron content of 11.2 mg/ml (41). After overnight incubation, culture medium was replaced by fresh medium containing SPIOs (0–150 μg Fe/ml). For labeling efficiency and viability studies, cells were incubated with SPIOs for 2, 4, 6, 16, and 24 h. For all other experiments, SPIOs were included in the culture medium for 16 and/or 24 h. Medium containing SPIOs was then discarded and cells were washed three times with phosphate-buffered saline (PBS) to remove free particles.
For longer term cultures, after 16 h of labeling standard medium containing SPIOs was replaced by WEM supplemented with 0.2 mg/ml bovine serum albumin (BSA), 20 μg/ml linoleic acid, 2 μg/ml linolenic acid, 2.45 μg/ml cAMP, 0.1 μM dexamethasone, 0.01 IU/ml prolactin, 0.06 ng/ml ethanolamine, 1 μg/ml glucagon (Sigma-Aldrich, UK), 9 ml/L insulin-transferrin-selenium-X 100× supplement (Gibco, UK), 20 ng/ml hepatocyte growth factor, 50 ng/ml epidermal growth factor (Peprotech, USA), 2 mM L-glutamine, 10 mM HEPES, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 3 μl/ml Fungizone® (Invitrogen, UK). Medium was replaced every 48 h during the first week and every 72 h thereafter (11).
Analysis of SPIOs Uptake
Prussian Blue Staining and Immunofluorescence
Uptake of SPIOs was visualized using Prussian blue (PB) staining and anti-dextran immunostaining. After labeling, cells were washed with PBS and detached according to Funaki et al. with some modifications (14). Briefly, cell cultures were incubated with PBS solution containing ethylene diamine tetraacetic acid (EDTA) [1.86 g/L of EDTA 2Na, 0.45 g/L of glucose, 2.38 g/L of soybean trypsin inhibitor (Sigma-Aldrich, UK) at pH 7.4] for 10 min at 37°C, washed twice with PBS, and incubated for a further 10 min with 0.05% collagenase solution containing 50 mg/L of DNase and 2 mM of CaCl2. Detached cells were collected, centrifuged at least twice for 3 min at 50 × g at 4°C, and resuspended in WEM. Cells were then transferred onto glass slides and fixed with 4% paraformaldehyde (PFA). For PB staining, cells were incubated for 20 min with 2% potassium ferrocyanide in 6% HCl. Slides were washed with PBS and counterstained with nuclear fast red (Sigma-Aldrich, UK).
Immunofluorescence staining for dextran was performed on cells cultured in collagen-coated chamber slides. After three washes with PBS, cells were fixed, permeabilized, and incubated with a FITC-conjugated mouse monoclonal anti-dextran antibody (StemCell Technologies, USA) at a 1:10 dilution for 1 h at room temperature (RT). Cells were mounted in Vectashield medium containing DAPI (4′-6-diamidino-2-phenylindole) nuclear counterstain (Vector Laboratories, USA). Slides were visualized using a Zeiss Axio-Imager Microscope equipped with an Axiovision 4.5 digital acquisition system.
Cellular Iron (Fe) Content
Intracellular iron uptake was determined using a colorimetric ferrozine-based assay (34). After labeling, cells were detached, washed with PBS, and lysed with 50 mM NaOH. One-hundred microliters of the cell lysate was mixed with 10 mM HCl (100 μl) and equal volumes of 1.4 M HCl and 4.5% (w/v) KMnO4 (100 μl), and incubated at 60°C for 2 h. Thirty microliters of iron detection reagent (6.5 mM ferrozine, 6.5 mM neocuproine, 2.5 M ammonium acetate, and 1 M ascorbic acid) (Sigma-Aldrich, UK) was added to each tube. Absorbance of samples was measured at 550 nm on a microplate reader (Dynex MRX, Dynex Technologies Ltd., UK) and compared to a standard curve prepared using FeCl3 (0–300 μM). Measurements were performed in duplicate and results were expressed as pg Fe/cell.
Viability and Function
Cytotoxicity assays were performed on hepatocytes seeded onto collagen-coated 96-well plates in eight replicates and repeated at least three times for each time point.
Sulphorhodamine (SRB) Cytotoxicity Assay
This assay was used to determine the effects of SPIOs on cell attachment (25). After incubation with SPIOs, cell cultures were washed with PBS and fixed with 50% trichloroacetic acid solution. Wells were rinsed with water and cells stained with 0.4% SRB solution. Excess of dye was removed by washing with 1% acetic acid. Protein-bound dye was solubilized by using Tris-base unbuffered solution. Plates were read at 550 nm using a microplate reader.
MTS Assay
Mitochondrial dehydrogenase activity of labeled hepatocytes was assessed using the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) (Promega, UK) according to the manufacturer's instructions. Absorbance of formazan produced was measured using a microplate reader at 490 nm. Results were expressed as percentage of controls.
Protein Synthesis
Incorporation of [14C]leucine was determined after labeling cells with SPIOs for different periods of time. Briefly, 200 μl of culture medium containing 0.1 μCi L-[U-14C] leucine (Amersham, UK) was added per well and plates were incubated for 24 h. Cells were then harvested onto glass fiber membranes using a cell harvester (FilterMate™, Packard Instruments, UK). Radioactivity was counted using a β-counter (MATRIX™ 9600 Direct β Counter, Packard Instruments, UK) and results were expressed as percentage of 14C incorporation of controls without SPIO labeling.
Specific Metabolic Functions
For the assays described below, cells were cultured in collagen-coated 24-well plates. Measurements were performed in duplicate, and repeated at least three times. Results were normalized against the protein content of each well (22).
Albumin and transferrin concentrations were measured in culture supernatants after 16 and 24 h of incubation with SPIOs using human albumin and transferrin enzyme-linked immunosorbent assay (ELISA) kits (Bethyl Laboratories, USA), according to the manufacturer's recommendations. Results were expressed as μg albumin or transferrin/mg cell protein. Urea formation was measured in the culture supernatants using the colorimetric QuantiChrom™ assay kit (BioAssay Systems, USA) following the manufacturer's protocol. Results were expressed as μg urea/mg cell protein. To evaluate long-term effects of labeling on hepatocyte specific functions, cells were labeled for 16 h with increasing concentrations of SPIOs. Albumin and urea concentrations in the supernatants were determined at days 4, 8, and 14 of culture. Twenty-four hours before collection of the supernatant, urea synthesis was stimulated with 2 mM of ammonium chloride.
Cytochrome P450 (CYP1A1/2) activity after 24 h of labeling was assessed using the ethoxyresorufin O-deethylation method (10). Briefly, 8 μM of 7-ethoxyresorufin substrate and 10 μM of dicumarol were added to each well and incubated at 37°C for 30 min. To include the formation of resorufin conjugates, culture supernatants were incubated with β-glucuronidase and arylsulfatase at 37°C for 2 h. The reaction was stopped by addition of ethanol and fluorescence was read at 530 nm excitation/590 nm emission, using a fluorescence microplate reader (Tecan Genios Pro). Results were expressed as pmol resorufin/min/mg cell protein.
Reactive Oxygen Species Formation
Intracellular formation of ROS was determined using 5-(and-6-)-choromethyl-2′,7′ dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Invitrogen, UK). Conversion of nonfluorescent CM-H2 DCFDA to green fluorescing CM-DCF indicates production of organic hydroperoxides that are formed after lipid peroxidation. After 16 h of labeling, cells were incubated with 10 μM of CM-H2DCFDA for 30 min at 37°C, washed with PBS, and harvested by scraping with a rubber policeman. A total of 10,000 cells were acquired on a FACSCalibur and analyzed using CellQuest software. The results were expressed as a percentage of the mean fluorescence intensity (MFI) of corresponding controls.
Hepatocyte Transplantation in Mice
Animals
Nonobese diabetic/severe combined immune deficiency (NOD-Scid) mice (Harlan, UK) of 6–8 weeks of age and weighing between 20 and 30 g were used as hepatocyte recipients. The animals were allowed to acclimatize for at least 1 week prior to HT. Food and water were available ad libitum. All procedures were in accordance with the UK Animals (Scientific) Procedures Act of 1986 and the ethical review process of King's College London.
Hepatocyte Labeling and Transplantation
Cells were prepared for transplantation by labeling with SPIOs at 50 μg Fe/ml for 16 h. Cells were washed three times with PBS and detached as described above. Detached cells were washed twice by centrifugation at 50 × g for 3 min, counted, and resuspended in Roswell Park Memorial Institute (RPMI)-1640 medium. Animals received 2.5 × 106 SPIO-labeled (n = 3) or nonlabeled (n = 3) human hepatocytes in 500 μl of medium into the spleen. A control group (n = 3) consisted of intrasplenic vehicle injection of RPMI-1640 medium alone. Animals were anesthetized by isoflurane inhalation. The lower pole of the spleen was isolated via a subcostal incision. The cell suspension was injected using a 25-gauge needle with hemostasis secured by ligation of the tip of the spleen. After 2 h, the animals were anesthetized with isoflurane and perfused through the heart with heparinized saline (5,000 IU/L), followed by perfusion of the whole animal with 4% (PFA) in PBS to fix the tissues. The livers were evaluated by both MRI of the mice and liver tissue analysis.
Liver Histology
Transplanted human cells were detected on 5-μm-thick, PFA-fixed, paraffin-embedded tissue sections using human serum albumin immunohistochemistry and PB staining for iron as described above.
Relaxivity Measurements and Magnetic Resonance Imaging (MRI)
Based on the results of the cytotoxicity assays, hepatocytes were labeled for 16 h with 0–50 μg Fe/ml of SPIOs and then scanned (20,000 cells/μl) to determine the optimal contrast agent concentration for MRI detection. To determine the MRI detection threshold hepatocytes were labeled with 50 μg Fe/ml of SPIOs. Nonlabeled cells (20,000/μl) and different densities of labeled cells (20–20,000/μl) isolated from the same donor tissue were resuspended in liquid 6% gelatin (w/v) (Sigma-Aldrich, UK) and transferred to PCR tubes. The mixture was allowed to solidify rapidly at 4°C to prevent clumping of the cells in the gelatin. In order to stabilize the tubes and also to avoid artifacts from the surrounding air during the MRI scanning, the tubes were placed in a 4% agarose phantom. All MRI experiments were performed on a 7-T 30-cm-bore horizontal Varian magnetic resonance system (Varian Inc., UK) using a custom-made quadrature coil driven by VNMRJ 2.2.b software. A multislice spin-echo imaging sequence was used to acquire T2-weighted images [repetition time (TR) = 2,000 ms, 8 echo times = 10, 20, 30, 40, 50, 60, 70, 80 ms, 2 averages, field of view (FOV) = 50 × 50 mm, slice thickness = 2 mm, matrix size = 256 × 256]. Relaxivity measurements were based on these images that were used to calculate T2 maps using VNMRJ 2.2.b.
For the MRI of the mice, a multislice spin-echo imaging sequence was acquired for the calculation of T2 maps (TR = 2,800 ms, 8 echo times = 10, 20, 30, 40, 50, 60, 70, 80 ms, 4 averages). T2*-weighted images were acquired using a multislice gradient-echo sequence (TR = 500, flip angle = 15°, 6 averages). For the animal experiments, the FOV was 30 × 30 mm, with 0.6-mm-thick slices, and a 192 × 192 matrix size.
Statistical Analysis
Results are presented as mean ± SEM. Differences between labeled and nonlabeled (control) cells were tested using either the unpaired Student's t-test or ANOVA with repeated measurements, with comparisons between groups performed by the least significant difference (LSD) test. A value of p ≤ 0.05 was considered statistically significant.
Results
Analysis of Labeling Efficiency
Incubation of hepatocytes with SPIOs resulted in incorporation of the contrast agent, as demonstrated by PB staining for iron (Fig. 1A), but the uptake was heterogeneous. Nonlabeled hepatocytes did not show any stainable iron (Fig. 1B). Incorporation of the contrast agent nanoparticles was confirmed by anti-dextran immunofluorescent staining (Fig. 1C). Intracellular iron concentrations increased in a time- and dose-dependent manner after incubation with SPIOs up to 18.5 ± 3.4 pg Fe/cell (Fig. 1D). Hepatocytes were labeled with 25 μg Fe/ml of SPIOs for 16 h and then maintained in culture for up to 8 days. At day 8, intracellular iron concentration was 7.9 ± 0.7 pg Fe/cell, showing retention of the label.
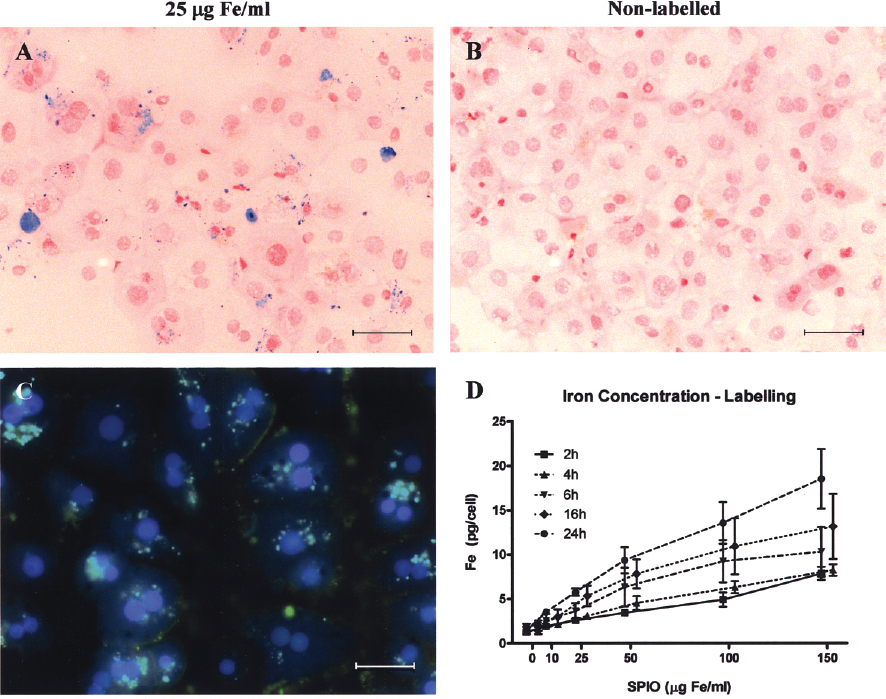
SPIOs uptake by primary human hepatocytes occurs in a nonsaturable manner. (A) Prussian blue staining at day 1 of hepatocytes labeled with 25 μg Fe/ml for 16 h observed at light microscopy as blue granules in the cytoplasm, and (B) nonlabeled cells (controls). (C) Iron particle uptake was confirmed by immunofluorescence using a FITC-conjugated antibody against the dextran coating of the contrast agent (green) and DAPI for nuclear counterstain (blue). Scale bars: 30 μm. (D) Graph showing a dose- and time-dependent increase in intracellular iron content, and that saturation did not occur.
Effects of Iron Incorporation on Cell Viability
After incubation with SPIOs, the number of attached cells measured by the SRB assay at different time intervals was similar to that in controls (Fig. 2A). The mitochondrial activity was increased in cells labeled for up to 6 h (p < 0.05). A significant decrease in formazan formation from MTS was only observed after cell incubation with 150 μg Fe/ml for ≥16 h (16 h = 82.2 ± 6.0%, p< 0.001; 24 h = 86.0 ±3.1%, p < 0.01) compared to the corresponding controls (Fig. 2B). However, a significant dose- and time-dependent reduction in [14C]leucine incorporation by hepatocytes was evident after SPIO labeling, decreasing up to 60% after 24 h of labeling with 150 μg Fe/ml compared to controls (p < 0.05) (Fig. 2C).
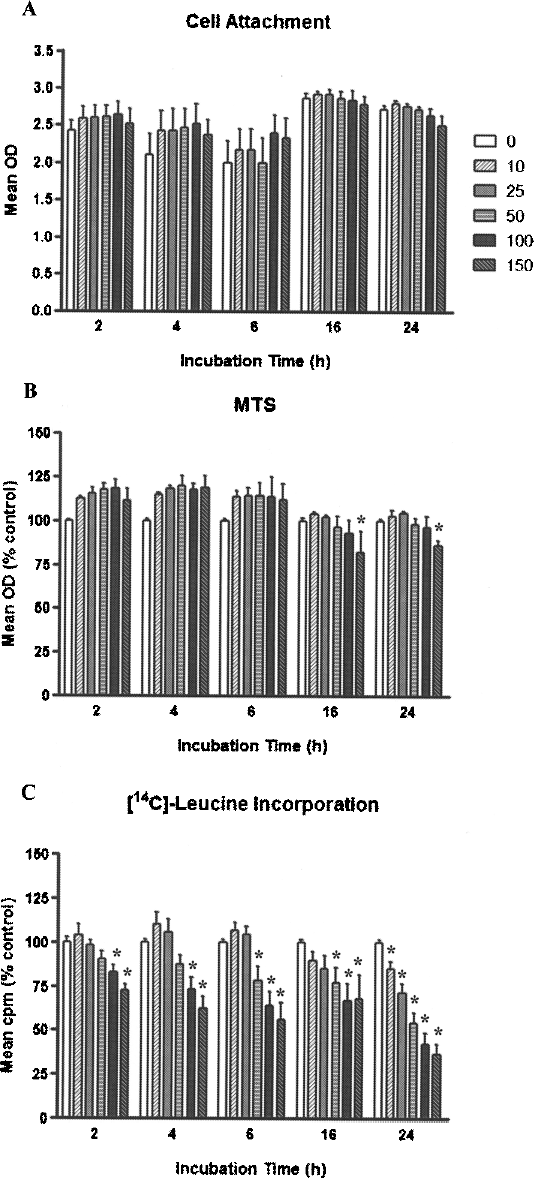
Viability and function of labeled hepatocytes. (A) SPIO labeling of human hepatocytes did not affect cell attachment, measured by the SRB assay, at any concentration or time point studied. (B) Similarly, mitochondrial activity in the MTS assay was comparable to control levels, except for a significant decrease after 24 h of labeling with 150 μg Fe/ml. (C) In contrast, incorporation of [14C]leucine by SPIO-labeled cells was significantly reduced with increased SPIOs concentration and incubation time after labeling (*p < 0.05 compared with corresponding controls).
Effects on Liver-Specific Function
After 16 h of incubation with SPIOs, albumin production in labeled cells was comparable to corresponding controls (Fig. 3A). Longer incubation times (24 h) with SPIOs resulted in a decrease in albumin production (50 μg Fe/ml = 4.2 + 0.4, p < 0.01; 100 μg Fe/ml = 3.3 + 0.4, p < 0.001; 150 μg Fe/ml = 3.3 + 0.4 μg albumin/mg cell protein, p < 0.001) compared to control (6.6 ± 1.0 μg albumin/mg cell protein). Longer term cell cultures after SPIO labeling demonstrated a dose-dependent decrease in albumin production (p < 0.05) at day 4 with subsequent recovery at day 8 and 14 in cell cultures labeled with <50 μg Fe/ml to values comparable to control levels (Fig. 3B).

Short-and long-term effects of labeling on hepatocytes. (A) Albumin production measured by ELISA after 16 h of labeling with SPIOs was not affected by labeling at any of the concentrations studied, but there was a significant decrease after 24 h of labeling with SPIOs concentrations >50 μg Fe/ml (*p < 0.05). (B) For long-term studies, cells were labeled for 16 h with increasing concentrations of SPIOs and maintained in culture for further 14 days. At day 4 there was a significant reduction in albumin secretion into the supernatant, with higher concentrations of SPIOs. There was a subsequent recovery at day 8 and 14 with values comparable to control levels in cell cultures labeled with <50 μg Fe/ml. (C) No significant difference in urea production was observed between controls and SPIO-labeled cells in any of the concentrations studied up to 24 h. (D) There was a progressive decrease in urea synthesis during the culture period in all groups studied. (E) The ability of cells to produce and secrete transferrin into the supernatant after 16 and 24 h of incubation with SPIOs was comparable to control cells. (F) Drug metabolizing capacity measured by the CYP1A1/2 activity was not affected after 24 h of labeling.
No significant difference in urea production between SPIO-labeled cells and controls was observed on day 1 (Fig. 3C). Subsequently, urea synthesis progressively decreased during the culture period up to 14 days in all groups studied, particularly in cells labeled with ≥100 μg Fe/ml, although this reduction was not statistically significant (Fig. 3D).
The ability of labeled cells to synthesize transferrin was not significantly affected after 16 and 24 h of incubation with SPIOs compared to controls (Fig. 3E). CYP1A1/2 activity after 24 h of labeling was similar to control levels at all concentrations studied (Fig. 3F).
ROS Production
An increase in intracellular unbound iron is known to induce generation of a wide range of ROS, based on the Fenton and Haber-Weiss reactions. No significant changes in ROS production were observed in cells within 16 h after labeling (10 μg Fe/ml = 105.0 ± 14.4; 25 μg Fe/ml = 99.6 ± 16.3; 50 μg Fe/ml = 101.6 ± 16.2; 100 μg Fe/ml = 107.3 ± 20.7; 150 μg Fe/ml = 107.7 ± 24.2% of control MFI) compared to controls.
In Vitro MRI and Detection Threshold
SPIO-labeled hepatocytes produced a dose-dependent decrease in T2 relaxivity compared to nonlabeled cells. In order to determine the detection threshold of this method and to establish a correlation between number of hepatocytes and change in T2 relaxivity, increasing numbers of SPIO-labeled cells (from 20 to 20,000 cells/μl) were imaged at day 1 and 4 after labeling. T2 relaxivity was dependent on the number of labeled cells. At day 1, concentrations of 2,000, 4,000, and 20,000 SPIO-labeled cells/μl induced a decrease of 14%, 48%, and 84%, respectively, on T2 relaxivity compared to nonlabeled cells (Fig. 4A). At day 4, the changes in T2 relaxation times were still evident and concentrations of 2,000 labeled cells resulted in a T2 relaxivity change of 35% (Fig. 4B).

Relaxometry of cells labeled with 50 μg Fe/ml for 16 h, embedded in 6% gelatin, and scanned using a 7-T MRI system. Depending on the cell concentrations, SPIO-labeled cells shortened T2 relaxation times by (A) 14%, 48%, and 84% (for 2,000, 4,000, and 20,000 cells/μl, respectively) compared to corresponding control cells at day 1 after labeling, and (B) by 35%, 51%, and 84% at day 4.
MRI Detection of Transplanted Cells and Histological Assessment
SPIO-labeled human hepatocytes were transplanted intrasplenically into NOD-Scid mice and animals were scanned 2 h after the procedure. Labeled cells clearly induced a decrease in signal intensity in the liver on T2*-weighted images, showing that labeled cells translocated throughout the liver and can be detected by MRI (Fig. 5A). To determine the contribution of SPIOs to the detection of transplanted human hepatocytes, animals were transplanted with nonlabeled hepatocytes and vehicle only injection. In these animals, transplanted cells did not change the MRI signal (Fig. 5B, C). Human albumin immunoperoxidase and PB staining performed on liver sections of mice transplanted with SPIO-labeled cells showed the presence of clusters of cells expressing human albumin, which also stained for iron located mainly within the portal spaces (Fig. 5D). Stained cells had the typical morphology of hepatocytes. Liver of animals transplanted with nonlabeled hepatocytes showed cells expressing human albumin, which did not stain for iron (Fig. 5E). Cells expressing human albumin were absent in livers transplanted with vehicle-alone injection (Fig. 5F). Human liver and mouse liver were used as positive and negative controls, respectively, for human albumin immunoperoxidase staining (Fig. 5G, H).
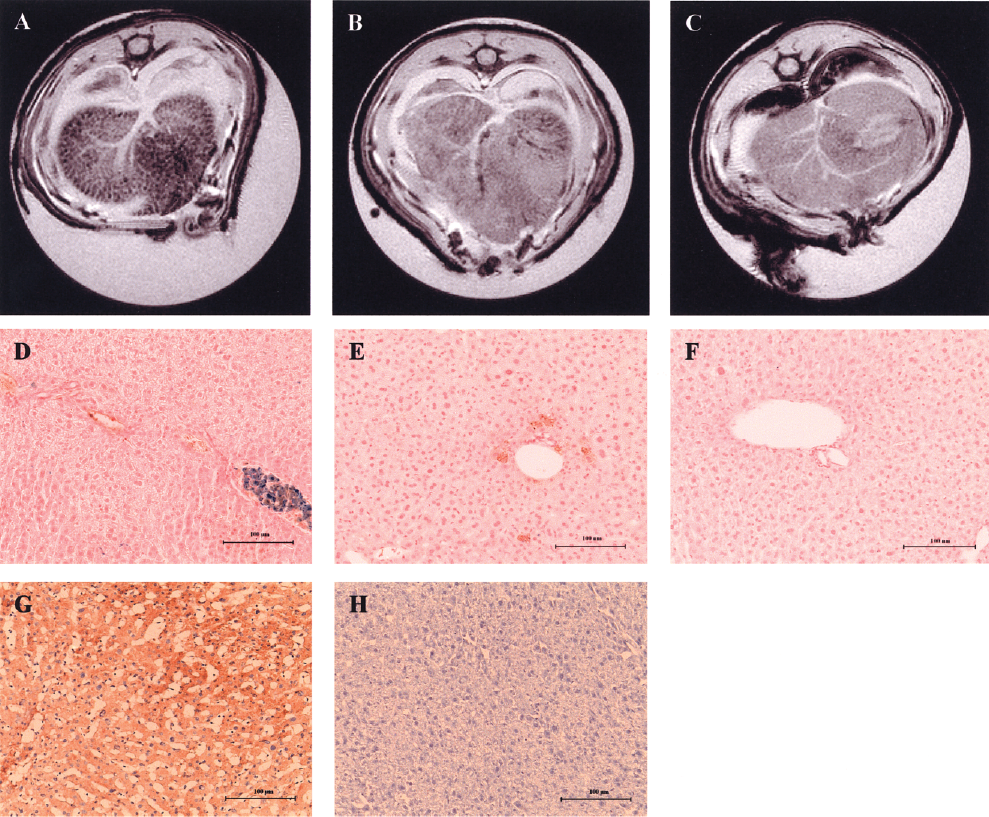
MRI of NOD-Scid mouse livers and validation with liver histology. (A) Hepatocyte labeling with SPIOs induced areas of hypointense signal on T2*-weighted MR images, whereas (B) nonlabeled hepatocytes and (C) vehicle injection of RPMI-1640 medium did not change the signal intensity. (D) Human albumin immunoperoxidase (brown) and PB staining (blue) shows clusters of labeled cells consistent with hepatocytes still present in the portal spaces. No stainable iron was detected by PB staining on liver sections of animals that have received (E) nonlabeled cells or (F) vehicle injection only. Expression of human albumin by immunoperoxidase staining on (G) human (positive control) and (H) mouse liver (negative control). Sections were counterstained with haematoxylin. Scale bars: 100 μm.
Discussion
This study has shown that human hepatocytes incorporate clinically approved dextran-coated SPIOs, which could be detected by MRI after hepatocyte transplantation in mice. SPIOs had no adverse effects on cell attachment and minimal effects on mitochondrial metabolic activity, with a slight decrease seen only in those cell cultures incubated with higher concentrations (150 μg Fe/ml) of SPIOs. Specific liver functions, such as urea and transferrin synthesis, and CYP1A1/2 activity were preserved after labeling. However, longer term culture of the cells with SPIOs demonstrated that, at higher concentrations, labeling affected albumin and urea production.
There are previous reports of adverse effects of SPIOs on other types of cells in culture. Human fibroblasts exposed to 50 μg/ml of SPIOs for 48 h showed an increase in apoptosis and cell death, aberrations in cell morphology, and decreased cell motility (3). Incubation with iron oxides with an anionic surface coating resulted in dose-dependent cytotoxicity and cell detachment, increased cytoskeletal disruption, and reduced ability of pheochromocytoma cells to appropriately respond to nerve growth factor (31). Other adverse effects reported included inhibition of proliferation (5) and impairment of differentiation of mesenchymal stem cells into chondrogenic lineage (21). Hussain et al. (18) presented data on the toxicity of various metal oxide nanoparticles on immortalized rat hepatocyte cultures and showed a cytotoxic effect of iron oxides at 250 μg/ml dose. Assessing potential ill effects of contrast agents in vitro is essential as minor changes might be indicative of agents interfering with cellular repair processes as has previously been demonstrated for a gadolinium-based agent (6,26).
The mechanism of incorporation of SPIOs into cells is probably by fluid-phase endocytosis. Our results demonstrated that incorporation of SPIOs by liver cells was nonsaturable in the range of concentrations and incubation times tested. However, PB staining showed a heterogeneous SPIOs uptake with no stainable iron in some of the cells. Similar uptake patterns have been reported in other studies that used iron oxide particles to label cells (28).
The retention of the contrast agent within the cells, along with preservation of cellular viability and function, are essential to reliably monitor the fate of cells in vivo for a period of time after transplantation. In this respect iron could be detected in labeled cells by MRI after 4 days and was still quantifiable in the cells after 8 days of culture. However, it is possible that iron did not remain in the form of nanoparticles in the cells. The core of SPIOs consists of magnetite (Fe3O4) and maghemite (γFe2O3) stabilized by a dextran coating (8). Previous reports in other cells have shown that the low pH of endosomes/lysosomes, as well as the presence of endogenous iron chelates, can solubilize the iron core within a few days, releasing ferric iron [Fe(III)] into the cytoplasm (2,36). The colorimetric ferrozine-based assay used in this study cannot differentiate the form of iron, as Fe(III) is reduced into ferrous iron [Fe(II)] with ascorbic acid. Intracellular iron concentration levels are controlled by both cellular transferrin receptors and cytoplasmic ferritin. In the presence of H2O2, free iron catalyzes formation of ROS. When ROS levels exceed the antioxidant capacity of the cells, it can cause cell damage through oxidation of proteins, peroxidation of lipid membranes, and DNA damage (20,30). In this study, exposure of cells to SPIOs for 16 h did not result in an increase in ROS formation, suggesting that nanoparticles were still intact at this point.
SPIOs create magnetic field inhomogeneity and dephasing of the transverse relaxation, causing a decrease in signal intensity on T2- and T2*-weighted MRI, which gives the hypointense images. However, if nanoparticles are broken down and free Fe(III) is released into the cytoplasm an increase in the signal intensity would be expected on T2-weighted images, as Fe(III) is paramagnetic and shortens the T1 relaxation time (2). This would be a limitation for longer term studies after transplantation of labeled cells. In addition, Endorem® is RES-specific contrast agent, so that particles released from damaged hepatocytes will be taken up by Kupffer cells, initially labeling these cells followed by rapid degradation of the SPIOs and return of the MRI signal intensity to normal levels (42). In the current study, tracking of SPIO-labeled human hepatocytes injected into the livers of NOD-Scid mice was performed in the first few hours of transplantation, to prove the principle that transplanted cells could be detected in the recipient liver by MRI. Further time would be needed for engraftment of cells into the liver parenchyma to occur. A significant fraction of transplanted hepatocytes in the liver sinusoids and portal tracts are cleared by Kupffer cells (19), which would also lead to transitory labeling of Kupffer cells as described above.
Larger micrometer-sized iron oxide particles (MPIOs) are also being investigated as a promising MRI contrast agent for cellular imaging (16). This contrast agent consists of a magnetite core in an inert styrene-divinyl benzene polymer shell. Shapiro et al. reported spontaneous hepatocyte uptake of MPIOs and in vivo single cell detection after intrasplenic injection of labeled hepatocytes in a murine model (35). Primary human hepatocytes also showed a fast uptake of MPIOs in culture, with no effects on viability or function, while being readily detected by MRI (33). However, these particles are nonbiodegradable and thus may present problems for translation to clinical use. The use of clinically approved SPIOs for imaging transplanted hepatocytes, as in the current study, will make it easier for clinical application. The biodegradable properties of these particles make this approach safe, with no expected long-term side effects, although there is the disadvantage of possible loss of detectability of the labeled cells. This technique should allow detection of cell biodistribution and/or loss of cells at an early stage after transplantation to allow appropriate intervention.
In summary, this study has defined conditions for labeling human hepatocytes in culture with SPIOs at sufficient levels for MRI detection, without significant deleterious effects. Labeled hepatocytes transplanted into the liver via the spleen were clearly detected in mice on T2*-weighted MRI sequences soon after transplantation. With further experiments to optimize the conditions for labeling human hepatocytes, particularly to obtain more uniform labeling of cells, it should be possible to develop this technique for use in hepatocyte transplantation in patients with liver disease.
Footnotes
Acknowledgments
These authors would like to thank the Liver Transplant and Hepatobiliary Surgery teams and the Liver Pathology Service at King's College Hospital for their cooperation in obtaining liver tissues. J.P. was supported by an Alex Mowat PhD Studentship. M.M. is supported by an EU FP VII grant dedicated to cellular MR imaging (201842-ENCITE).