
Abstract
Umbilical cord blood (UCB) is recognized as an enriched source of endothelial progenitor cells (EPCs) with potential therapeutic value. Because cryopreservation is the only reliable method for long-term storage of UCB cells, the clinical application of EPCs depends on our ability to acquire them from cryopreserved samples; however, the feasibility of doing so remains unclear. In this study we demonstrate that EPCs can be isolated from cryopreserved UCB-derived mononuclear cells (MNCs). The number of outgrowth EPC colonies that emerged in culture from cryopreserved samples was similar to that obtained from fresh UCB. Furthermore, EPCs obtained from cryopreserved MNCs were phenotypically and functionally indistinguishable from freshly isolated ones, including the ability to form blood vessels in vivo. Our results eliminate the necessity of performing cell isolation procedures ahead of future clinical needs and suggest that EPCs derived from cryopreserved UCB may be suitable for EPC-related therapies.
Keywords
Introduction
Umbilical cord blood (UCB) is an established source of hematopoietic stem cells (HSCs) used in the treatment of hematologic, oncologic, immunologic, and metabolic diseases (3,4,7–9,11,18,19). As a result, UCB is now routinely collected, frozen, and stored in liquid nitrogen worldwide in anticipation of its future clinical use. In addition to HSCs, examination of UCB cellular components has uncovered other stem/progenitor cells with potential therapeutic value. For example, endothelial progenitor cells (EPCs), first identified in adult peripheral blood (1,12) but present in significantly higher numbers in UCB (10,16,17), form vascular networks in vivo (2,15,20,22), a characteristic that has created motivation for developing new EPC-based vascularization therapies.
Although numerous laboratories already work with EPCs isolated from fresh mononuclear cells (MNCs), successful EPC isolation from cryopreserved UCB has not been reported. Current protocols follow early recommendations to plate freshly obtained MNCs as the most reliable method for obtaining outgrowth EPC colonies in culture (5,14). However, the development of successful clinical therapies will likely depend on our ability to acquire functional EPCs from cryopreserved UCB. Here we demonstrate for the first time that EPCs can be isolated and culture expanded from cryopreserved MNCs. Moreover, EPCs from cryopreserved blood are phenotypically and mechanistically indistinguishable from their freshly isolated counterparts and retain their in vivo vasculogenic properties. These results suggest that EPCs obtained from banked UCB samples may be suitable for future clinical applications.
Materials and Methods
Isolation of EPCs From Fresh and Cryopreserved Cord Blood-Derived MNCs
Human UCB samples (n = 3; 30 ml each) were obtained from cesarean sections and ex utero at the Brigham and Women's Hospital in accordance with an Institutional Review Board-approved protocol, which waived the need for informed consent due to the samples being from discarded specimens. The complete MNC fraction was obtained (14,16) and divided into three groups, which were used as follows: a) plated in culture (herein referred to as fresh), b) cryopreserved (10% DMSO in FBS) and stored in liquid nitrogen for 1 month before plating (referred to as cryo-1), and c) cryopreserved for 4 months before plating (cryo-4). Each of these groups contained the same amount of MNCs. All MNCs were seeded on 1% gelatin-coated plates using EPC medium (EGM-2 without hydrocortisone supplemented with 20% FBS and 1× glutamine-penicillin-streptomycin) with 15% autologous plasma (16). Unbound cells were removed at 48 h. The number of endothelial-like colonies (≥ 50 cells) was counted under an inverted microscope at 2 weeks. At confluence, cells were purified using CD31-coated beads as previously described (14,16). Purified EPCs are referred to as fresh-EPCs and cryo-EPCs depending on whether they originated from fresh or cryopreserved MNCs, respectively.
Phenotypic Characterization of EPCs
EPCs between passages 3 and 6 were characterized by standard flow cytometry (CD31, CD90, and CD45), indirect immunostaining (CD31, vWF, and VE-Cadherin), and Western blot (CD31 and α-SMA) as previously shown (15,16). For leukocyte adhesion and adhesion molecule assays, EPC monolayers were challenged with or without 10 ng/ml of tumor necrosis factor-α (TNF-α) for 5 h. Afterwards, the leukemia cell line HL-60 (2 × 106 cells) was added and incubated at 4°C on a rocking platform for 45 min (6). Bound leukocytes were visualized and quantified using a phase contrast microscope and ImageJ analysis software. Additionally, leukocyte adhesion molecules were analyzed by flow cytometry using PE-conjugated antibodies against human E-selectin and ICAM-1 (6,16). Migration assays were carried out in 8-μm trans-wells. Cells were added to the upper chamber and incubated in DMEM containing 2% FBS with or without 50 ng/ml human VEGF-A (added to the lower chamber) for 16 h. The number of migrated cells was determined in triplicate by counting the cells present on the lower surface of the membrane after DAPI staining using a fluorescence microscope. Karyotyping was carried out from passage 8 cultures by G-banded analysis at the Brigham and Women's cytogenetic core facility (Boston, MA).
In Vivo Vasculogenic Properties of EPCs
Vasculogenesis was evaluated in vivo using our xenograft model as previously described (15). Briefly, EPCs and bone marrow-derived mesenchymal progenitor cells (MPCs) (2 × 106 total; 40:60 EPC/MPC ratio) were resuspended in 200 μl of Matrigel and subcutaneously injected into 6-week-old male athymic nu/nu mice (n = 4). Implants were harvested at 7 days, and histological (H&E) and immunohistochemical (human CD31) analyses were performed as previously shown (15). Microvessel density (vessels/mm2) was reported as the average number of erythrocyte-filled microvessels in sections from the middle of the implants (15).
Microscopy
Phase microscopy images were taken with a Nikon Eclipse TE300 inverted microscope using Spot Advance 3.5.9 software and a 4×/0.1 objective lens. Fluorescent images were taken with a Leica TCS SP2 Acousto-Optical Beam Splitter confocal system equipped with a DMIRE2 inverted microscope using a 63×/1.4 oil objective lens. Histology images were taken with a Primo Start Zeiss microscope equipped with an AxioCam MRc5 camera using a 40×/0.65 objective lens.
Statistical Analysis
The data were expressed as means ± SEM. Unless otherwise stated, all p-values reported were generated by individually comparing fresh-EPC and cryo-EPC groups using two-tailed Student's unpaired t-tests. Additionally, multiple comparisons were performed where appropriate by one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison tests. Values of p < 0.05 were considered statistically significant.
Results
MNCs isolated from fresh UCB samples were cryopreserved by our group and stored for one (cryo-1) or four (cryo-4) months in liquid nitrogen; in all cases, EPC colonies (identified by cobblestone morphology) emerged in culture with similar size, frequency, and time of appearance to those obtained from fresh UCB MNCs (Fig. 1). After quantification of all EPC colonies present at day 14, no statistically significant differences were found between fresh, cryo-1, and cryo-4 groups of MNCs (p = 0.7548, one-way ANOVA) obtained from three independent UCB preparations (Fig. 1B). Additionally, we cryopreserved UCB-derived MNCs using an alternative cryopreservation medium (90% autologous plasma, 10% DMSO, and 1% dextran-40) for 1 month in an effort to mimic clinical cryopreservation methods and found no statistical difference when compared to the number of colonies obtained with FBS-based cryopreservation medium (data not shown). Next, EPCs from each group were purified by CD31 selection and characterized using standard assays. Indirect immunofluorescent staining showed that cryo-EPCs expressed the endothelial (EC) markers CD31, VE-cadherin, and vWF (Fig. 2A). While CD31 and VE-cadherin were localized at the cell-cell borders, vWF was expressed in a punctuate pattern in the cytoplasm. Together, these were clear indications of an EC phenotype. Flow cytometry showed uniform expression of CD31 and absence of CD90 and CD45, confirming that neither cryo-EPCs nor fresh-EPCs were contaminated with mesenchymal or hematopoietic cells (Fig. 2B). Western blot analysis confirmed expression of CD31 and absence of α-SMA, a marker of perivascular and mesenchymal cells (Fig. 2C). In addition, both cryo-EPCs and fresh-EPCs were equally responsive to VEGF as shown by migration assay (p = 0.4802) (Fig. 2D). Finally, cells cultured for eight passages were karyotypically stable (Fig. 2E depicts representative sorted karyograms of cryo-EPCs showing a normal male karyotype of 46,XY), suggesting that acquisition of EPCs from cryopreserved MNCs does not increase the risk of chromosomal abnormalities.

Outgrowth EPC colonies from fresh and cryopreserved MNCs. Freshly isolated UCB-derived MNCs were divided into three groups: 1) noncryopreserved (fresh), 2) cryopreserved and stored in liquid nitrogen for 1 month (cryo-1), and 3) cryopreserved for 4 months (cryo-4). All MNCs were seeded on 1% gelatin-coated plates using EPC-medium. (A) Representative EC-like cell colonies obtained from fresh and cryo-1 MNCs at 2 weeks (white arrowheads mark colonies borders). Scale bar: 0.5 mm. (B) The number of colonies (≥ 50 cells) from each MNC group was counted at 2 weeks from three independent UCB samples (n = 3). p-Values were generated by one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison tests.

Phenotypic characterization of EPCs isolated from cryopreserved MNCs. (A) Indirect immunofluorescence of cryo-EPCs grown in a confluent monolayer showing positive staining for CD31, VE-cadherin, and vWF. IgG control depicted in inset. Scale bar: 50 μm. (B) Flow cytometry analysis of fresh-EPCs and cryo-EPCs for EC marker CD31, mesenchymal marker CD90, and hematopoietic marker CD45. Black line histograms represent cells stained with fluorescent antibodies. Isotype-matched controls are overlaid in solid gray histograms. (C) Western blot analysis of fresh-EPCs and cryo-EPCs for EC marker CD31 and mesenchymal/perivascular marker α-SMA. Human bone marrow-derived MPCs served as control. β-Tubulin accounted for loading control. (D) Migration assays were carried out by challenging fresh-EPCs and cryo-EPCs (n = 3 each) with or without VEGF-A for 16 h. Each bar represents the mean number of EPCs migrated ± SEM from three independent UCB samples. p-Values were generated by individually comparing fresh-EPC and cryo-EPC groups using two-tailed Student's unpaired t-tests. (E) Representative sorted karyograms of cryo-EPCs showing a normal male karyotype of 46,XY. Karyotyping was carried out from passage 8 cultures by G-banded analysis.
We also evaluated functional properties of cryo-EPCs. First, we tested whether cryo-EPCs upregulated leukocyte adhesion molecules in response to the inflammatory cytokine TNF-α. As expected, E-selectin was low to undetectable and ICAM-1 was low (though detectable) in the untreated cryo-EPCs. Both adhesion molecules were markedly upregulated after 5 h of incubation with TNF-α (Fig. 3A). Quantification by flow cytometry revealed that upregulation of E-selectin and ICAM-1 was the same in cryo-EPCs and fresh-EPCs (p = 0.4171 and p = 0.1417, respectively) (Fig. 3A, B). Also, this upregulation resulted in increased binding of HL-60 leukocytes (Fig. 3C). Quantitative analysis of leukocyte adhesion after TNF-α treatment showed no statistically significant differences between cryo-EPCs and fresh-EPCs from the three independent cord blood preparations (p = 0.7775) (Fig. 3D). Equivalent upregulation of adhesion molecules (a functional property of ECs) in cryo-EPCs and fresh-EPCs suggests that cryopreservation does not impair physiological proinflammatory response in EPCs.
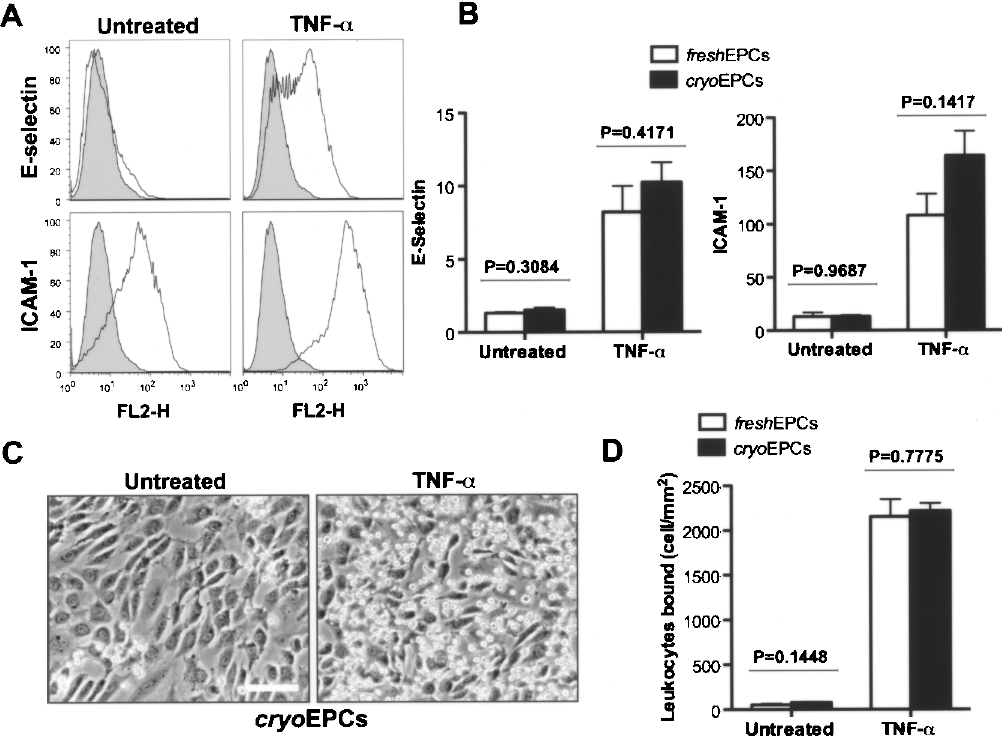
Proinflammatory properties of EPCs isolated from cryopreserved MNCs. (A) In vitro upregulation of leukocyte adhesion molecules E-selectin and ICAM-1 in response to TNF-α. Representative flow cytometry histograms are depicted for cryo-EPCs. Black line histograms represent cells stained with fluorescent antibodies against adhesion molecules, while solid gray histograms correspond to the isotype-matched controls. (B) Flow cytometry quantification of E-selectin and ICAM-1 upregulation in response to TNF-α in fresh-EPCs and cryo-EPCs. Each bar represents the mean fluorescent value of the entire EPC population ± SEM from three independent UCB samples. (C) Representative phase contrast pictures of confluent cryo-EPCs showing an increased number of bound HL-60 leukocytes after TNF-α treatment (right) compared to untreated control (left). Scale bar: 100 μm. (D) Quantification of bound leukocytes. Each bar represents the mean number of HL-60 cells bound to the EPC monolayer ± SEM from three independent UCB samples. p-Values were generated by individually comparing fresh-EPC and cryo-EPC groups using two-tailed Student's unpaired t-tests.
Finally, we evaluated the in vivo vasculogenic abilities of cryo-EPCs using our assay of human EPCs and MPCs coimplanted into immune-deficient mice. As already reported with fresh-EPCs (15), histological examination of cryo-EPC explants revealed extensive networks of microvessels filled with murine erythrocytes at day 7 (Fig. 4A). These engineered microvessels stained positive for human CD31, confirming the structural participation of EPCs in blood vessel formation (Fig. 4A), while murine capillaries were negative for human CD31 (black arrowheads). Quantification of average microvessel densities at day 7 revealed no statistically significant differences between implants prepared with cryo-EPCs and fresh-EPCs (p > 0.5, t -tests; n = 4 mice) (Fig. 4B). When comparing all groups together from three independent UBC preparations, there were no statistically significant differences (p = 0.6077, one-way ANOVA). Carefully examination of vessels showed no histological evidence of thrombosis (e.g., platelet aggregates and uniform fibrin deposit). This lack of thrombosis was true for vessels engineered with both fresh-EPCs and cryo-EPCs, suggesting that both EPCs have the expected antithrombotic properties of endothelial cells in vivo.
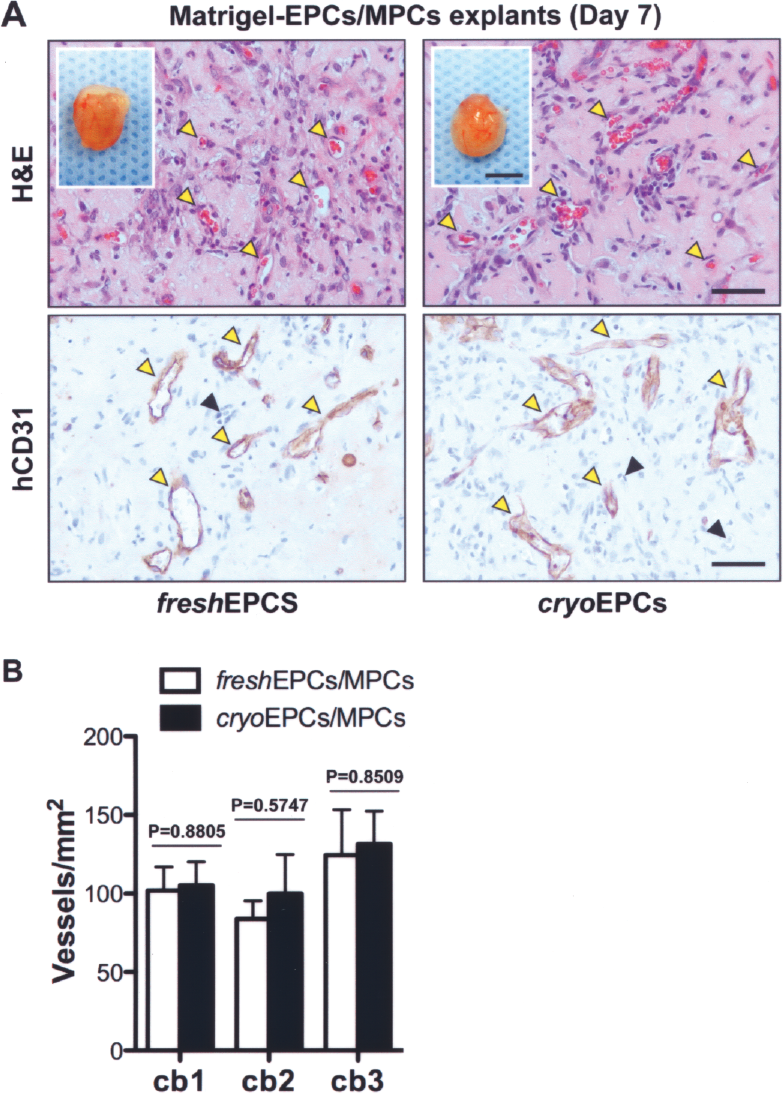
In vivo vasculogenic ability of EPCs isolated from cryopreserved MNCs. In vivo vasculogenic properties were evaluated by subcutaneous coimplantation of MPCs with either fresh-EPCs or cryo-EPCs into 6-week-old nude mice. Both fresh-EPCs and cryo-EPCs were acquired from three independent UCB samples (cb1–cb3). (A) Macroscopic views of explants at day 7 are depicted in insets. Scale bar: 5 mm. H&E staining at day 7 (top panels) revealed the presence of numerous blood vessels containing murine erythrocytes (yellow arrowheads). Immunohistochemistry (bottom panels) showed that engineered microvessels stained positive for human CD31 (yellow arrowheads), while murine capillaries (black arrowheads) did not. All images are representative of implants harvested from four different mice (yellow arrowheads indicate blood vessels) Scale bar: 50 μm. (B) Microvessel densities of implants harvested at day 7 were quantified by counting lumenal structures containing erythrocytes. For each UCB sample evaluated (cb1–cb3), bars represent the mean microvessel density determined from four replicate implants (one mouse each) ± SEM. p-Values were generated by individually comparing fresh-EPC and cryo-EPC groups using two-tailed Student's unpaired t-tests.
Discussion
The discovery of EPCs in peripheral blood created an exciting opportunity to noninvasively obtain large quantities of autologous ECs for either therapeutic vascularization or tissue engineering. However, the process of obtaining blood-derived EPCs with bona fide blood vessel-forming ability has not been straightforward due to the low concentration of circulating EPCs relative to leukocytes and the lack of a unique set of distinctive EPC markers. For these reasons, the isolation of circulating EPCs by flow cytometry or other immunological techniques is still a challenging problem. As a result, the most successful way to isolate EPCs is based on methods similar to those originally reported for endothelial outgrowth colonies from peripheral blood (12). First, MNCs are collected from fresh blood and plated onto collagen-coated plates in endothelial-specific growth media (10,12,22), and then, EC-like colonies emerge from the adherent cell population after 1–3 weeks in culture (16,22). Despite the success of this procedure, the methodology is limited by the requirement of freshly collected blood.
The clinical use of UCB will likely rely on cord blood cryopreservation and storage in specialized cell banks. Therefore, the development of UCB-derived EPC therapies will involve acquisition of functional EPCs from cryopreserved UCB. In this study, we demonstrate for the first time the feasibility of obtaining EPCs from cryopreserved UCB MNCs. Following the methodology described above, we obtained similar numbers of EPC colonies regardless of whether the original MNCs were freshly obtained or cryopreserved. We also evaluated the phenotype of the cryo-EPCs using current standards in the field. Taken together, there were no differences between fresh and cryopreserved EPCs at these endpoints: 1) expression of EC markers CD31, VE-cadherin, and vWF, 2) absence of expression of CD45 (hematopoietic marker), CD90, and α-SMA (mesenchymal markers), 3) responsiveness to VEGF-A (migration), 4) upregulation of leukocyte adhesion molecules upon exposure to an inflammatory cytokine (TNF-α), 5) increased adhesion of leukocytes in response to TNF-α, 6) blood vessel-forming ability in vivo, and 7) chromosomal stability after extensive (passage 8) expansion in culture, a mandatory requirement for any future cell-based therapy. Therefore, in addition to finding a robust EC phenotype, our characterization revealed no phenotypical or functional differences between cryo-EPCs and fresh-EPCs. It is of note that our findings do not corroborate a recent study reporting that cryopreservation of UCB impairs the ability to obtain EPCs in culture (13). However, these differences may be explained by examination of the EPC isolation protocols used: while Lu et al. (13) relied on CD34+ cell sorting of MNCs, we directly plated MNCs in culture, avoiding additional processing of the thawed cells. More recently, Vanneaux et al. reported that cryopreservation and thawing significantly altered the capacity to generate and expand EPCs (21). However, we did not find statistically significant differences when both the fresh and the cryopreserved MNCs were obtained from the same UCB samples.
Our studies demonstrate that EPCs with blood vessel-forming abilities can be isolated from cryopreserved UCB-derived MNCs and cryo-EPCs are phenotypically and functionally indistinguishable from fresh-EPCs. Even though our experiments were performed using UCB samples that were cryopreserved by our group in the laboratory, the possibility of obtaining EPCs from frozen UCB cells constitutes a step forward in the development of EPC-based clinical therapies. The use of frozen UCB cells eliminates the necessity of performing expensive cell isolation procedures ahead of potential clinical needs and reduces the dependency on freshly collected UCB, which is especially convenient for laboratories not located near clinical facilities.
Footnotes
Acknowledgments
We would like to thank Dr. Sule Cataltepe at Brigham and Women's Hospital for provision of UCB samples. Histology and karyotyping were supported by the Specialized Research Pathology Cores, Longwood Facility of the Dana-Farber/Harvard Cancer Center (P30 CA06516). This work was partially supported by NIH grant K99EB009096-01A1 to J.M.-M. and K99CA140708-0109 to A.C.D.