
Abstract
Umbilical cord blood (CB) represents a main source of circulating endothelial progenitor cells (cEPCs). In view of their clinical use, in either the autologous or allogeneic setting, cEPCs should likely be expanded from CB kept frozen in CB banks. In this study, we compared the expansion, functional features, senescence pattern over culture, and in vivo angiogenic potential of cEPCs isolated from fresh or cryopreserved CB (cryoCB). cEPCs could be isolated in only 59% of cryoCB compared to 94% for fresh CB, while CB units were matched in terms of initial volume, nucleated and CD34+ cell number. Moreover, the number of endothelial colony-forming cells was significantly decreased when using cryoCB. Once cEPCs culture was established, the proliferation, migration, tube formation, and acetylated-LDL uptake potentials were similar in both groups. In addition, cEPCs derived from cryoCB displayed the same senescence status and telomeres length as that of cEPCs derived from fresh CB. Karyotypic aberrations were found in cells obtained from both fresh and cryoCB. In vivo, in a hind limb ischemia murine model, cEPCs from fresh and cryoCB were equally efficient to induce neovascularization. Thus, cEPCs isolated from cryoCB exhibited similar properties to those of fresh CB in vitro and in vivo. However, the low frequency of cEPCs colony formation after cryopreservation shed light on the need for specific freezing conditions adapted to cEPCs in view of their future clinical use.
Introduction
Until recently, neovascularization or formation of new blood vessels has been thought to result exclusively from proliferation and migration of preexisting endothelial cells, a process referred as to angiogenesis (7). Furthermore, vascularization, defined as in situ differentiation of vascular endothelial cells from endothelial precursor cells, was thought to occur only in the embryo during vascular development. It is nowadays established that circulating bone marrow-derived endothelial progenitor cells (cEPCs) are capable of homing to neovascularization sites, and proliferation and differentiation into endothelial cells (29,41).
cEPCs have been identified mainly in the mononuclear cell fraction of peripheral blood, leukapheresis products, and in umbilical cord blood (1,5). Recently cEPCs have been extensively studied as biomarkers to assess the risk of cardiovascular disease in human subjects. Indeed, a lower cEPCs count predicts severe impaired functions in several cardiovascular pathologies such as diabetes (12), hypercholesterolemia (8), hypertension (38,45), scleroderma (10,25), aging (17,45), cigarette smoking (23,32,45), and coronary artery disease (46). Transplantation of cEPCs into ischemic tissues thus emerged as a promising approach in the treatment of diseases with blood vessels disorders (6,22,28).
In the mouse, cEPCs injection led to improved neovascularization in hind limb ischemia (6,22,28). Ex vivo expanded cEPCs, isolated from peripheral blood mononuclear cells, can also incorporate into the foci of myocardial neovascularization (11,26), and intracoronary infusion of peripheral blood or bone marrow-derived progenitors in patients with acute myocardial infarction was associated with significant benefits in postinfarction remodeling (13,30,31,34,36,37,42,43).
For the two last decades, umbilical cord blood (CB) has served as a source of hematopoietic progenitor cells, which are used for the treatment of various high-risk hematological malignancies in children and adults. CB is also a treatment of choice in nonmalignant disorders and acquired or inherited bone marrow failure syndromes (39). CB banking for allogeneic hematopoietic stem cell transplantation has been established worldwide over the past, with cryopreservation methods allowing maintenance of viability over years (15).
Although the impact of freezing on hematopoietic stem cells has been extensively studied (4,16,24,33,47), it has not been clearly determined whether or not these processes maintain other stem and progenitor cells as viable and functional. This question is of crucial importance because thousands of CB cells are collected, cryopreserved, and stored in anticipation of their potential therapeutic application in regenerative medicine. As an example, CB is a main source of cEPCs, which can be ex vivo expanded with a higher frequency and a robust proliferative potential compared to cEPCs from adult peripheral blood (19). CB thus represents a valuable source for the production of cEPCs to be used in the treatment of a wide range of cardiovascular diseases. To that end, in both the autologous or allogeneic setting, cEPCs should likely be expanded from cryopreserved CB (cryoCB) in CB banks. In this study, we aimed to compare the expansion potential of cEPCs isolated from fresh or cryoCB, together with their in vitro and in vivo functional properties.
Materials and Methods
Isolation and Culture Conditions of CB cEPCs
Human umbilical CB was collected from normal full-term deliveries after maternal informed consent according to approved institutional guidelines (Assistance Publique-Hôpitaux de Paris, Paris, France). Cryopreserved CB was maintained at a temperature below −150°C at the Saint-Louis Hospital Cord Blood Bank, Paris, France. Mononuclear cells (MNCs) obtained from fresh or thawed CB were isolated by centrifugation on Pancoll (Dutscher, Burmath, France), plated onto collagen type I-coated wells (Becton Dickinson, Le Pont de Claix, France), and maintained in endothelial basal medium-2 (EBM2) supplemented with EGM2-MV Single-Quots (Lonza, Ermerainville, France) at 37°C, under 5% CO2, in a humidified incubator. First media change was performed after 24 h to eliminate nonadherent cells and daily for 7 days. Media was then changed every 2 days. The outgrowth of endothelial colony-forming cells (ECFC), characterized by formation of a cluster of cobblestone-appearing cells, was assessed daily. Colonies were enumerated by visual inspection using an inverted microscope (Olympus, Lake Success, NY) under 40x magnification. To obtain cEPC-derived cells, ECFC were trypsinized, seeded at 10,000 cells/cm2, and expanded in EGM2-MV media.
Flow Cytometry Quantification and Phenotype Analysis
Mononuclear cells were enriched as previously described (3), using a Human RosetteSep Progenitor Enrichment kit (Stem Cell technologies, Canada). Cells were then subjected to triple labeling with anti-CD133-phycoerhytrin (PE, Miltenyi Biotec, Paris, France), anti-CD34-fluorescein isothiocyanate (FITC, Becton Dickinson), and biotinylated anti-VEGFR-2 (KDR) (Sigma-Aldrich, Saint Quentin Fallavier, France) antibodies. Viability was assessed with 7-AAD (Becton Dickinson). Immunophenotyping of cEPC-derived cells was assessed by using the following monoclonal antibodies: CD31-FITC, CD144-PE (Becton Dickinson), VEGFR2-APC (APC, R&D Systems, Minneapolis, MN), and von Willebrand Factor (vWF) (Dako, Trappes, France). For intracellular labeling, cells were permeabilized with phosphate-buffered saline (PBS)/saponin solution. Antibodies and matched isotype control were incubated for 30 min at 4°C. Data were acquired and analyzed on a five-parameter flow cytometer (FACScalibur, Becton Dickinson, San Jose, CA) with CellQuest software (Becton Dickinson).
Cell Proliferation Potential
At subconfluence, cells were detached and counted. The number of population doublings (PD) was calculated at each passage using the following formula: log2 (nf/no), where no is the initial and nf the final cell number at each passage. The cumulative population doubling was calculated as the sum of all previous PDs. Population doubling time (PDT) and cumulative population doubling time (CPDL) were calculated using the following formula: t/PD, where t is the interval of time of culture between each passage.
In Vitro Angiogenesis Assay
Capillary tube formation was performed by laying Matrigel (Becton Dickinson) into a 12-well plate. cEPC-derived cells were seeded on Matrigel (2 × 105 cells/well) and suspended in EGM2-MV medium for 18 h at 37°C with 5% CO2. Capillary tube formation in Matrigel was observed under an inverted microscope and kinetic of tube formation was assessed by taking pictures every 2 hours.
Wound-Healing Assay
Cell migration was analyzed by using the method described by Sato and Rifkin with modifications (40). One linear scar was drawn in a confluent cEPC-derived cells monolayer. A set of digital photographs was taken. Dishes were then incubated for 18 h in EGM2-MV medium and a second set of photographs was taken.
Ac-LDL Uptake
Incorporation analysis of acetylated low-density lipoprotein 488 (Ac-LDL 488; Invitrogen, Cergy-Pontoise, France) was performed to assess the incorporation ability of cEPCs. Cells were incubated with 15 mg/ml of Ac-LDL 488 in endothelial medium for 4 h at 37°C, washed three times, and mounted with Glycergel mounting medium (Dako). Ac-LDL uptake was observed under a fluorescence microscope and analyzed with Metamorph software.
TNF-α Activation
Cells were seeded at 2 × 104 cells/well and, once at confluence, stimulated with TNF-α (10 ng/ml) for 18 h. After incubation, cells were trypsinized and stained with ICAM-1 and VCAM-1 primary antibodies (R&D Systems) and goat anti-mouse secondary antibody (Invitrogen). Data were acquired and analyzed on a five-parameter flow cytometer (FACScalibur, Becton Dickinson).
Senescence-Associated (SA)-β-Galactosidase Assay
Cells were washed, fixed, and incubated overnight at 37°C with X-gal chromogenic substrate at pH 6.0 following the manufacturer's instructions. After staining, cells were analyzed on pictures of five randomly selected fields.
Telomere Length
Genomic DNA was prepared using a “High pure PCR template preparation kit” (Roche Diagnostics, Meylan, France). For this purpose, 2 μg DNA was digested at 37°C with restriction enzymes RsaI and HinfI (Roche Diagnostics). DNA fragments were separated by gel electrophoresis at 130 V on a 1% agarose gel and transferred to a nylon membrane. Membranes were washed twice in 2x SSC for 15 min at room temperature and hybridized with digoxigenin solution and telomeres oligonucleotides for 3 h at 42°C. Membranes were washed in 1x SSC, 0.1% SDS for 15 min at 50°C. Terminal restriction fragments (TRFs) length was determined by chemiluminescent detection.
RNA Extraction and Quantitative PCR
Cells were collected at passages 4, 7, and 8. Total RNA was extracted with RNeasy mini-kit (Qiagen, Hil-den, Germany) and cDNA prepared with the cDNA Ar-chive kit (Applied Biosystems, Foster City, CA). Quantitative PCR (qPCR) was performed with TaqMan® Gene Expression Assays on an ABI7000 (Applied Biosystems) in the presence of 10 ng of RNA. CD146, eNOS, KDR, ephrin-B4, Eph-B2, Alk-1, COUPTFII, and RPLP0 (ribosomal protein, endogenous control) expressions were assessed (predesigned assays, references respectively: Hs00163543_m1, Hs00174838_m1, Hs00819630_m1, Hs00167166_m1, Hs00187950_m1, Hs00174752_m1, Hs00176676_m1, Hs99999902_m1). The 2-ΔΔCt method was used and results expressed as gene expression fold increase relative to human umbilical vein endothelial cells (HUVECs).
Karyotype Analysis
cEPCs at 70% confluence in culture with EBMV2 medium were incubated with colchicine (4 μg/ml, Sigma-Aldrich) for 3 h. The cells were then trypsinized and incubated with hypotonic solution (0.075 M KCl). Metaphases were fixed with methanol/acetic acid (3:1, v/v). Cytogenetic analysis was performed using R banding procedure. Images were acquired with the MET-AFER system (METAsystems, Germany) on Zeiss microscope and chromosomes were classified with the IKAROS software (METAsystems, Germany). The results were reported according to International System for Human Cytogenetic Nomenclature (ISCN; 2005). A minimum of 20 metaphases were screened from each sample.
Hind Limb Ischemia Model
In vivo cEPCs angiogenesis potential was assessed in a NOD/SCID mice model of hind limb ischemia. NOD/SCID mice were purchased from The Villejuif Animal Experiment Laboratory. All experimental procedures were performed under French authorities' guidelines. Twelve 8-week-old female mice were anesthetized with a mix of ketamin and xylazine. The left proximal and distal parts of the femoral artery of the left leg were then ligated (6.0 silk suture, Ethicon, Issy-Les-Moulineaux, France) and the part between ligations was excised. Seven days after surgery, cEPC-derived cells expanded from fresh or cryoCB were intramuscularly injected in tibialis anterior of the ischemic limb (1 × 106 cells per animal). A PBS/albumin injection was used in the control group. Hind limb blood flow was measured using a scanner-laser Doppler (Laser Doppler Perfusion system, Perimed Periscan II). Average perfusion of ischemic and nonischemic limbs was determined before and after ischemia, and 34 days after ischemia. Blood flow-dependent changes in laser frequency were imaged using different colored pixels. Images were analyzed to quantify blood flow by using a blood perfusion analyze software (LPDIwin). Percentage of perfusion was expressed as the ratio of the ischemic to the nonischemic hind limb. Thirty-four days after surgery, animals were sacrificed. Muscles isolated from the ischemic hind limb were fixed in paraffin and stained with HES (hemaluneosine-saffron).
Statistical Analysis
For in vitro experiments, results were analyzed with Fisher test followed by the Student test. Data obtained from in vivo experiment were expressed as geometric means between the two groups, with the associated 95% confidence intervals (CI), and analyzed with a two-way analysis of variance (ANOVA) with p-values adjusted with the Bonferonni method. A value of p < 0.05 was considered as indicating significant difference.
Results
Isolation, Expansion, and Characterization of cEPCs Derived From Fresh and cryoCB
cEPCs were quantified as lin/7AAD/CD34+/CD133+/KDR+-positive cells in the mononuclear fraction of fresh (n = 17) and thawed (n = 17) CB. The number of cEPCs/1 × 106 viable mononuclear cells was significantly higher in thawed CB compared to that obtained in fresh CB (123.8 ± 103.8 and 18.6 ± 14, respectively, p < 0.05) (Fig. 1A), while the initial number of nucleated cells was similar in both conditions, as well as the CB volumes initially harvested. Despite that, the mean number of ECFC was dramatically reduced in the thawed CB group (3.5 ECFC for thawed CB vs. 13.9 ECFC for fresh CB, p = 0.02) (Fig. 1B), even if ECFC morphology was similar (Fig. 1C). The ECFC outgrowth potential was a mean of 7.6 days (range 5–14) in the cryoCB group, compared to 10.7 days (range 5–16) for fresh CB (p < 0.05). However, we were able to generate ECFC from only 59% of thawed CB whereas 94% of fresh CB efficiently gave rise to ECFC, thus demonstrating that cryopreservation and thawing significantly altered the capacity to generate and expand cEPCs.

Isolation and expansion characteristics of CB cEPC-derived cells. (A) cEPC quantification per 106 viable mononuclear cells (MNC) in fresh and cryoCB. cEPC number was significantly higher in thawed CB compared to fresh CB (p < 0.02). (B) Mean number of ECFC obtained from fresh and cryoCB. ECFC growth was observed after 5–22 days of culture. (C) Representative phase contrast microscopy of endothelial colony-forming cells (ECFC) obtained from either fresh or cryoCB (original magnification 40x). Scale bars: 200 μm. (D) Cell growth kinetics of cEPC-derived cells. Cumulative population doubling levels (CPDL) are shown for both fresh and cryoCB.
Because cryopreservation affects, to a great extent, the survival of human cEPCs, we next evaluated whether or not it also modifies other biological parameters such as immunophenotype, proliferation potential, and specific endothelial gene expression, and compared them to fresh cEPCs taken as a reference. In both cases, cEPC-derived cells displayed the same endothelial phenotype with high expression of CD31 (99 ± 0.05% and 98.8 ± 1.4%, respectively), KDR (85.4 ± 12% and 85.7 ± 19.5%, respectively), vWf (98 ± 1.9% and 93 ± 5.8%, respectively), and CD144 (45 ± 24% and 67.4 ± 29%, respectively). Similarly, once culture was established, cell growth kinetics was not significantly different between fresh and cryoCB, assessed by either population doubling or number of passages (Fig. 1D).
We next analyzed the expression of genes described to be specific of venous (ephrin-B4 and COUPTFII) or arterial (Eph-B2 and Alk-1) endothelial cells, as well as those commonly expressed in all endothelial cell types (eNOS and CD146). Gene expression was studied in cEPC-derived cells expanded from fresh or cryoCB at different passages (passage 4, 7, and 8), and compared to that of HUVEC, known to display a venous phenotype. The levels of expression were not significantly different between the two groups or between cells at early or late passages (Table 1). cEPC-derived cells expressed CD146 and eNOS endothelial genes at the same levels to that of HUVEC. Interestingly, cEPC-derived cells also expressed both the arterial and venous endothelium-specific genes, suggesting that CB cEPC-derived cells have no arterial and/or venous fate determination.
Quantitative PCR on CB cEPC-Derived Cells
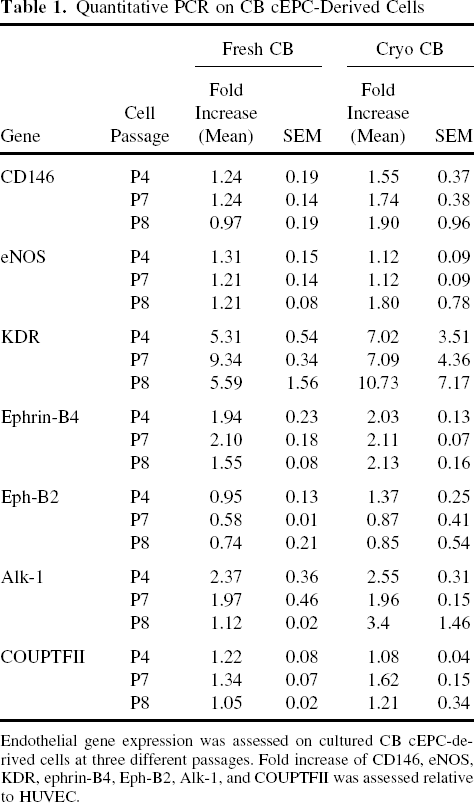
Endothelial gene expression was assessed on cultured CB cEPC-derived cells at three different passages. Fold increase of CD146, eNOS, KDR, ephrin-B4, Eph-B2, Alk-1, and COUPTFII was assessed relative to HUVEC.
Functional Characteristics of CB cEPC-Derived Cells
We first analyzed the ability of CB cEPC-derived cells to form capillary-like network in vitro. cEPC-derived cells from fresh or cryoCB were seeded in Matrigel and cultured during 18 h. As shown in Figure 2A and B, capillary-like structures were observed in both cases. Immunohistochemistry experiments in which cEPC-derived cells were labeled with anti-CD31 antibodies confirmed that these tubular vessels like structures were composed of endothelial cells (Fig. 2C, D).

In vitro functional characteristics of CB-cEPCs. cEPC-derived cells obtained from fresh (A, C) or cryopreserved (B, D) CB created capillary-like network on Matrigel after 18 h of culture. (A, B) Phase contrast microscopy; (C, D) CD31 staining (in red). Representative images of wound healing assay at T0 and after 24 h performed by using cEPC-derived cells from fresh (E, G) or cryoCB (F, H). cEPC-derived cells were able to uptake acetylated LDL coupled to 488 fluorochrome (I, fresh CB; J, cryoCB). Scale bars: 200 μm (A, B, G, H, I and J) and 100 μm (C, D, E and F).
Because migration capacity is one of the endothelial cell features, we ran an in vitro wound-healing assay using cEPC-derived cells expanded from fresh or cryoCB. In both experiments, the cells were shown to over-run the scars and to form a continuous monolayer (Fig. 2E-H). The time the cells took to migrate and cover the scar did not differ significantly among the two groups. This wound-healing model is not solely a migration model. It also involves cell proliferation at the wound border. To confirm that cell proliferation did not impact the results of this assay, cell growth kinetic was evaluated at the time the wound-healing assay was performed. Similar cell population doublings were observed between fresh and cryoCB-derived cells (data not shown). The ability to incorporate Ac-LDL has been described as another characteristic of endothelial cells. cEPC-derived cells expanded from fresh or cryoCB were incubated in the presence of Ac-LDL during 4 h. As shown in Figure 2I and J, the Ac-LDL uptake was demonstrated to be efficient in both cases and similar to that observed in HUVECs (not shown).
Phenotypically, and under standard culture conditions, cEPC-derived cells express low levels of ICAM-1 and do not express VCAM-1 molecule. However, these levels of expression are increased in response to TNF-α stimulation. We incubated cEPC-derived cells in the presence of 10 ng/ml TNF-α and analyzed the expression of these two adhesion molecules at the membrane surface. After stimulation, an upregulation of both ICAM-1 and VCAM-1 was observed, with greatest effects seen with VCAM-1 (Fig. 3). The expression fold induction was similar between cEPC-derived cells isolated from fresh or cryoCB, with a median of induction of 3.2 and 3.9 for ICAM-1, respectively, and of 24.6 and 17.2 for VCAM-1, respectively.

TNF-α-induced ICAM and VCAM expression. cEPC-derived cells from fresh or cryoCB were cultured in the presence or absence of TNF-α (10 ng/ml) for 18 h. ICAM-1 and VCAM-1 expression was analyzed by flow cytometry. Histograms are shown where thin dotted line represents isotype control, bold dotted line expression before stimulation, and solid line expression after TNF-α stimulation.
Lastly, we determined if some differences existed between cells grown from fresh or cryoCB regarding senescence and telomere length. cEPC-derived cells were harvested at various culture times and stained with SA-β-galactosidase, a marker of cell senescence. Whatever the number of cell passages considered (early or late passages, i.e., passage 2 or 10), no differences were detected between cells grown from fresh or cryoCB in this assay (Fig. 4A). The cell viability, tested in parallel, was also similar in both conditions: 96.1 ± 1.2% and 96 ± 2% at passage 2 and 10, respectively, for fresh CB, and 95.8±1.4% and 96.5±1.2% at similar passages for cryoCB (Fig. 4B). Telomere length was measured from passage 2 to passage 14, on cells expanded from cryopreserved or fresh CB. While a slight and expected decrease of telomere length was observed at the highest passages (Fig. 4C), no significant differences could be evidenced between the two groups, thus suggesting that cryopreservation and thawing did not influence cell growth and survival, once the cells are obtained.

CB cEPC-derived cells senescence pattern. (A) Representative photographs of senescence assay based on β-galactosidase activity on cEPC-derived cells at passage 2 and 10. cEPC-derived cells from fresh CB and from cryoCB. Senescent cells appear in blue (scale bars: 100 μm). (B) Histograms represents cell viability, assessed by 7-AAD, at passage 2 (P2) and 10 (P10), for fresh CB (white bars) and cryoCB (black bars). (C) Telomere length analysis on cells from fresh or cryoCB, performed at various culture time (passage 2 to 14).
Karyotype Analysis of CB cEPC-Derived Cells
Karyotype analysis was performed on six samples of CB cEPC-derived cells (fresh CB, n = 3; cryoCB, n = 3). As shown in Table 2, tetraploid metaphases were detected in three samples (sample 1, fresh CB; samples 2 and 3, cryoCB), and a trisomy of chromosome 11 in sample 3 (cryoCB). In the three remaining samples, a normal karyotype was found.
Karyotype Analysis of CB cEPC-Derived Cells From Fresh (Samples 1, 4, 5) or cryoCB (Samples 2, 3, 6)

At least 20 metaphases were analyzed after R-banding. The results are reported according to International System for Human Cytogenetic Nomenclature (ISCN; 2005).
In Vivo Hind Limb Ischemia
In vivo neovascularization studies were performed in the NOD/SCID hind limb ischemia model. cEPC-derived cells isolated from fresh or cryoCB (1 × 106 per mouse), or placebo (PBS/albumin buffer) were intramuscularly given via tibialis injection 7 days after femoral artery ligation. Laser Doppler blood flow measurements were taken from both the injured and uninjured legs of all animals. As shown in Figure 5A, blood perfusion was dramatically decreased at day 1 in all groups, thus triggering ischemia of the operated legs. At day 34, higher perfusion ratios were noted in mice injected with cEPC-derived cells from either fresh or cryoCB compared to mice receiving placebo treatment. HES coloration showed necrotic areas in control PBS-treated mice whereas hind limb muscles exhibited a normal muscular fiber organization in the animals transplanted with cEPC-derived cells (Fig. 5A). The percentage of perfusion of ischemic limb was completely restored at day 34 for mice receiving cEPC-derived cells compared to the levels measured before femoral artery ligation (Fig. 5B). In contrast, perfusion ratios in the control remained low at day 34 (61%) compared with 98.3% and 103.6% in the groups that received cEPC-derived cells from fresh or cryoCB, respectively (p < 0.05 in both cases compared to control).

In vivo comparison of EPC-derived cells from fresh and cryoCB. (A) Representative laser Doppler images of blood flow in the hind limbs before and after 1 and 34 days after ischemia. Injection of cEPC-derived cells 7 days after ischemia enhances reperfusion in ischemic limb and leads to a faster reparation in treated mice. Histologic HES coloration, performed 34 days after ischemia, showed better recovery of muscles injected with cEPC-derived cells compared to control. Scale bars: 200 μm. (B) Percentage of perfusion obtained from ischemic-to-nonischemic limb ratios. The measurements were performed before surgery (-1), 1 day, and 34 days after surgery. *p < 0.001 versus day 1, #p < 0.05 versus control.
Discussion
Endothelial progenitor cells derived from frozen CB units holds promise as cellular therapy or tissue engineering for numerous human vascular diseases (20,35). For years it has been demonstrated that CB represents a useful alternate source of cEPCs compared to peripheral blood. A high number of cEPCs could actually be isolated from CB, with increased proliferative potential compared to their adult counterpart (18,19) and better in vivo potential to form vessels (2). Given the important cell number required for therapeutic approaches, in vitro expansion is a prerequisite for cEPC-derived cell transplantation. To that end, and both in the autologous or allogeneic setting, cEPCs will be expanded from cryoCB.
Worldwide, at least in public CB banks, the CB cryopreservation process aims at ensuring long-term storage of hematopoietic stem cells, mainly with the view of allogeneic transplant for hematological disorders. However, it has not been clearly established whether these conditions allow isolating and expanding other stem or progenitor cell populations, or if their functional capacities are preserved after the thawing procedure. By using a CD34+ magnetic cell sorting for EPCs isolation and culture, Lu et al. (27) showed that they were unable to generate endothelial-like cells from cryopreserved CB. This was found to be mainly related to increased apoptosis of CD34+ cells after thawing.
In our study, we compare isolation, expansion, and in vitro and in vivo functional characteristics of cEPC-derived from fresh or cryoCB. Consistent with previous reports (2,9,14,18,21), we succeed in quantifying, isolating, and expanding cEPCs from almost all tested fresh CB. Conversely, cEPCs could be obtained in less than 60% of cryoCB, while these were of an initial volume of more than 70 ml and thawed in the same conditions as those used in the allogeneic transplantation setting. Moreover, the number of ECFCs obtained after CB thawing was significantly reduced compared to that of fresh CB, thus demonstrating that, even when cryopreserved with a standardized and validated procedure, thawed CB does not allowed efficiently obtaining cEPCs. These results were furthermore obtained where cEPCs count was significantly higher after CB thawing.
One could hypothesize that cryopreservation and thawing process of the whole CB led to an “enrichment” of the cEPCs fraction, mostly by inducing cell death of the more mature mononuclear cells and red cell lysis. Indeed, it has already been demonstrated that cEPCs could be isolated and differentiated into endothelial cells from frozen umbilical CB mononuclear cells (21). In that case, mononuclear cells were preliminarily isolated from CB and stored at −196°C before inducing cEPCs. However, even if the number of cEPCs was shown to be higher in thawed CB, the fact that the number of ECFCs was reduced tends to demonstrate that cEPCs were less functional than that obtained from fresh CB.
Once ECFCs could be obtained from cryoCB, the growth kinetics and proliferation potential of cEPCs and cEPC-derived cells were similar to that of cells expanded from fresh CB. At this stage, CB thawing did not seem to have any deleterious effect because no significant alteration of in vitro cEPCs properties was observed. Thus, cEPCs and cEPC-derived cells from fresh or cryoCB expressed the same membrane antigen that features these two cell types. The qPCR results we presented in Table 1 showed that cEPC-derived cells expressed both arterial and venous genes. Indeed, these cells expressed COUPTFII and ephrin-B4 venous genes, but also expressed arterial genes such as Alk-1. cEPC-derived cells thus displayed an hybrid arterial-venous phenotype.
In vitro functional properties, such as tube formation, migration, and Ac-LDL uptake potentials were shown to be similar in the two groups. ICAM and VCAM molecules expression before and after stimulation with TNF-α revealed no difference between cells expanded from fresh or cryoCB, nor with expression of these molecules on HUVEC. Taken together, these in vitro results demonstrated that cEPCs and cEPC-derived cells from human fresh or cryoCB shared similar functional properties. However, we also showed that, even using CB of high initial volume and cell number, the yield of cEPC-derived colonies was relatively low after cryopreservation and thawing. We thought these observations to be of interest in the setting of storing CB for future stem cell-based therapy (44). Furthermore, these data highlight the decision that has to be made regarding the storage of poor-quality or low-yield cord collections, especially in the autologous setting for which the criteria of CB quality and storage are still to date far from those internationally required for allogeneic CB banking. In these conditions, it is very likely that the yield of cEPC-derived endothelial cells will be very poor. Whether the frequency of cEPCs in frozen/thawed CB is representative of other stem cells has to be established. Because cEPC-derived colonies are relatively easy to obtain, this test may represent a useful quality control assay for CB banks.
Recently, the numerous advantages of CB over peripheral blood regarding cEPCs and their potential applications have been tempered by the observations made by Corselli et al. reporting chromosomal abnormalities in CB cEPC-derived cells (9). Even if cEPCs functional features were not altered by these abnormalities, and if none of the treated animals developed tumors, these data should be taken into consideration at least in the setting of future clinical application. In our hands, some cEPC-derived cells also harbored cytogenetic abnormalities; these data were obtained for cryoCB as well as for fresh ones, thus suggesting that the cryopreservation and thawing process likely does not increase the incidence of cytogenetic alterations. At the time the animals were sacrificed, we did not evidence any tumor formation, similar to that was reported by Corselli et al. (9). Lastly, we addressed the question of whether or not cryopreservation and thawing modified cell senescence and telomere length, two points that should be of importance if long-term expansion is required for therapeutic approaches. It is important to note that, despite the difficulties to generate cEPCs from some cryoCB, cell senescence assessed by SA-β-galactosidase staining was equivalent to that of cells isolated from fresh CB. Telomere length, determined at different culture time points, was also shown to be equivalent in the two conditions, thus demonstrating that cryopreservation does not led to deleterious cell modifications.
Because the in vitro studies cannot definitely predict cell functionality in vivo, we compared the potency of cEPC-derived cells from cryopreserved and fresh CB in a murine model of hind limb ischemia. We observed that cells expanded from both conditions equally and significantly increased blood reperfusion in the ischemic leg compared to the control group. These data are consistent with those reported by Finney et al. using the same animal model to compare neovascularization induced by cEPCs obtained from either bone marrow or CB (14). Histologic section analyses, performed on biopsies obtained at day 34, confirmed the neovascularization process by laser Doppler and showed muscular healing in animals treated with cEPC-derived cells. In this vascular injury model, we thus demonstrated that cEPC-derived cells obtained after CB thawing have equivalent efficacy in revascularization than cells from fresh CB.
In this study, we showed that cEPCs isolated from cryoCB exhibited similar in vitro and in vivo functional properties as those expanded from fresh CB. However, the relatively low frequency with which we successfully generated cEPCs after thawing has to be taken into consideration in view of their future clinical use. Actually, if cEPC-derived cells' therapeutic effect in the hind limb ischemia mice model gave similar results with fresh and cryoCB, we demonstrated that the number of ECFCs obtained after thawing was significantly reduced compared to that of fresh CB. As a matter of fact, the expansion process to be done for obtaining a sufficient cell number after CB thawing will be prolonged, with the risk, for example, of promoting chromosomal abnormalities. Improvements of specific freezing conditions are thus mandatory in order to optimize cEPCs recovery after CB thawing. However, it must not be forgotten that, to date, the vast majority of CB units are used in the setting of hematological diseases, and that thawing allows obtaining high hematopoietic stem cells recovery. Because these conditions could likely not be modified based upon cEPCs, an option might be to freeze cEPC-derived cells, beforehand expanded from part or whole fresh CB. On a clinical point of view, this strategy likely will not be reasonable for treating genetic diseases in an autologous setting because CB cEPC-derived cells could harbor genetic abnormalities. However, developing allogeneic CB cEPC-derived cell banks could be of use for proposing new therapeutic approaches for patients with ischemic injury.
Footnotes
Acknowledgment
This work was supported by a grant from the Agence Nationale de la Recherche (project “Eurocord Lab” No. ANR-07-RIB-005-02).