
Abstract
The double stranded Deoxyribonucleic acid (DNA) is stated as one of the idyllic targets for cancer and other various diseases. The in-depth investigation of DNA-drug interaction plays a crucial role for the recognition of drug mechanism action as well as in advance scheming of more proficient drugs with minor aftermath. Imidazoles and molecules possessing them are well known for their antimicrobial action and also possess different therapeutic properties. With the intention to improve a computational protocol towards the development of novel improvised antimicrobial agent, di-cationic molecules. Primarily, four diarylfuran derivatives having varied substituted groups have been proposed, analysed and compared for antimicrobial potential by studying their binding strength and steady complex formation with DNA. Molecular docking analysis was used to forecast the binding mode involved for DNA-drug complex and molecular dynamics was employed for studying the kinetics of interaction. The docking investigation revealed AT rich region binding for all the proposed ligands which is the preferred location for DNA minor groove binders establishing Mol-1 and Mol-3 as lead molecules. The stability of drug-DNA complexes was inferred from an analysis of the data from Molecular dynamics (MD) analysis, which showed that ligands remained coupled to the preferred binding locations of DNA without experiencing any significant changes in the minor groove.
Introduction
The biopolymer, Deoxyribonucleic acid (DNA) is considered as one of the outstanding targets for various diseases owing to its role as a source of genetic information [1]. Primarily small drug molecules bind to DNA and hinder with transcription and replication of DNA and eventually retard the cellular growth. The small molecule/drug implicates non-covalent mode of action with DNA namely intercalation, groove binding and electrostatic interactions. For the therapy of different microbial infections DNA groove binding is the ideal target [2]. Depending upon the width of DNA groove, binding is classified in two types i.e., minor (12Å) and major (22Å) and the diverse forces involved here are electrostatic, van der Waal’s, hydrophobic, and hydrogen bonding forces. The sequence specificity is quite crucial tool while studying the drug-DNA interaction. Owing to the higher electrostatic potential and narrower/deeper grooves, minor grooves elucidates AT region selectivity over GC [3, 4]. The DNA minor groove with alternating A and T offers favorable van der Waal’s interactions between the drug and DNA instead of guanine base rich GC-rich region. Small molecules having crescent shape corresponding to the DNA minor groove, numerous aromatic rings have the ability to form hydrogen bonds favors minor groove binding due to their cationic nature. The spine of hydration along the DNA minor groove is disrupted by the lipophilic aromatic rings. Studies involving drug-DNA complex formation are significant and in vogue presently since it could direct research toward more logical and target-oriented drug design. In the same line, theoretical analysis of intermolecular connection between drug/small molecules is of utmost importance. The dicationic molecules; Aromatic diamidines is well-known DNA minor grove binder and is shown to exhibit antimicrobial potency post binding to AT-rich regions [5]. Although having a broad-spectrum antimicrobial activity of Aromatic diamidines merely pentamidine has been found to be of substantial use clinically. However, Pentamidine could only be used against African trypanosomiasis, antimony-resistant leishmaniasis & AIDs-associated Pneumocystis carinii and is orally inactive and display problems of hypotension, dysglycemia in conjunction with renal and hepatic toxicity due to its metabolite’s toxicity [6–10]. Therefore, there is a continuous focus for the further development of improvised antimicrobial agents. In the present research work, minor groove binders, dicationic bis aryl derivatives labeled as Mol-1, Mol-2, Mol-3 and Mol-4 are selected from the literature [11], followed by a DNA sequence (PDB Id: 195D). Previous researches have shown similar DNA sequences to be better minor groove binders [12–18]. The literature suggested them to be potent antimicrobial agents and therefore in order to develop a computational protocol for the enhancement of existing and towards the search of novel antimicrobial agents, this study was performed [19]. For this research, molecular modelling methods like geometry optimization, molecule docking, and molecular dynamics simulation were applied [20]. The ligands were initially geometrically tuned to achieve a minimum potential to obtain electrostatically optimized chemical structure. The molecular docking is the simplest computational method for the determination of superlative docking site and an equivalent binding energy for Ligand-DNA interactions [14]. Following geometry optimization, ligands were docked to the chosen DNA sequence to identify potential binding sites, after which the matching site binding energy was also retrieved. According to the literature, better the corresponding docked position; the lower the site binding energy must be [21]. Post ligand geometry optimization and molecular docking, coarse-grained molecular dynamics simulations were carried out to drug-DNA complexes for 20 ns. The achieved outcomes strongly confirmed the interaction and stability of drug-DNA complexes are time-dependent and directed towards the conformational stability of DNA with ligands [13, 22].
Computational methodology
The proposed lead molecules (Mol-1, Mol-2, Mol-3 and Mol-4) were chosen from the previous research data ascribing them as potent antimicrobial agent [11]. The DNA sequence; 195D was acquired from the protein data bank [23, 24]. Before initializing the docking calculations, water molecules from DNA sequence were eliminated using UCSF Chimera, and certain visualizations were carried out using Discovery Studio software package [25, 26]. The geometry optimization of proposed ligands was performed using Gaussian 09 software [18] through B3LYP hybrid functional at 6-31G** level of theory [27]. QM charges were determined on each atom and molecules with assigned charges were later docked to the 195D DNA sequence. Further, to determine the binding site of potent inhibitors in the vicinity of target molecules molecular docking was carried out using Autodock4 software using Lamarckian Genetic Algorithm (LGA) [12, 28]. Gasteiger charges were supplemented to each drug-DNA complex by means of Autodock Tools (ADT). Afterwards, a grid box with varying sizes along the three coordinate axes was created for each drug-DNA complex encasing each macromolecule complex. After a 20 LGA with maximum cycle of 2500000 energy evaluations for each drug-DNA complex, least binding affinity docked pose was mined out and aligned with DNA for further investigations. We then carried out the molecular dynamics simulations of all the docked posed DNA-ligand complexes. It is an advanced computational technique to study the conformational changes in biomolecular systems with respect to time [29]. We used GROMACS 5.1.1 software to carry out the molecular dynamics in the current work [30]. We deployed the similar parameters as described in our previous work and carried out simulations in the present work as well [12, 31–36]. All the generated graphs were plotted using XMgrace software [37]. We further quantified our estimations of DNA-ligand interaction energies through carrying out free energy calculations. We used similar protocols as mentioned in one of our previous works [12, 13], and carried out the calculations. However, in this work we took snapshots at every 2 ns for the entire 20 ns trajectory and calculated components of the interaction energies for each snapshot. We also analyzed the per residue energy decomposition in order to view the highly stiff and flexible zones of the DNA during the simulations. The obtained results are discussed in detail in the forthcoming sections.
Results and Discussion
DNA minor groove is one of the most fascinating targets for cancer treatment and nearly all the developed therapeutic drugs reported involve interaction with DNA. The minor groove binder small molecules comprise high sequence specificity and greater binding affinity compared to intercalator binding. The compelling and broad-spectrum antimicrobial activity, selective DNA affinity of di-cationic, furamidine and related molecules has encouraged us for the further development in this area [38, 39]. For the drug development computational methodology for the analysis of receptor ligand interactions has gained momentum in the pharmaceutical industry. The major reason for this is non-requirement of costly experimental tools for these research analyses and henceforth they can be employed for complementing the experimental research. The current work is intended for the identification, assessment of binding affinity, structural stability of new di-cationic scaffold-based ligands through computational methodology. The optimized chemical structures of all selected ligands are depicted in Figure 1. We first executed the DFT calculations for the optimization and estimation of the geometry of ligands with DNA. This was followed by molecular docking analysis to predict the orientation of ligand within the DNA and study their interactions. Later, MD simulations for all the four ligands are carried out for 20 ns and results are stated in terms of variables such as Energy Variations, alteration in Radius of Gyration, Deviation in Number of Hydrogen Bonds, Root Mean Square Deviation and Root Mean Square Fluctuation. By the data provided by these variables, microstructural information such as the position and solvation of the DNA with proposed ligands can be analysed.
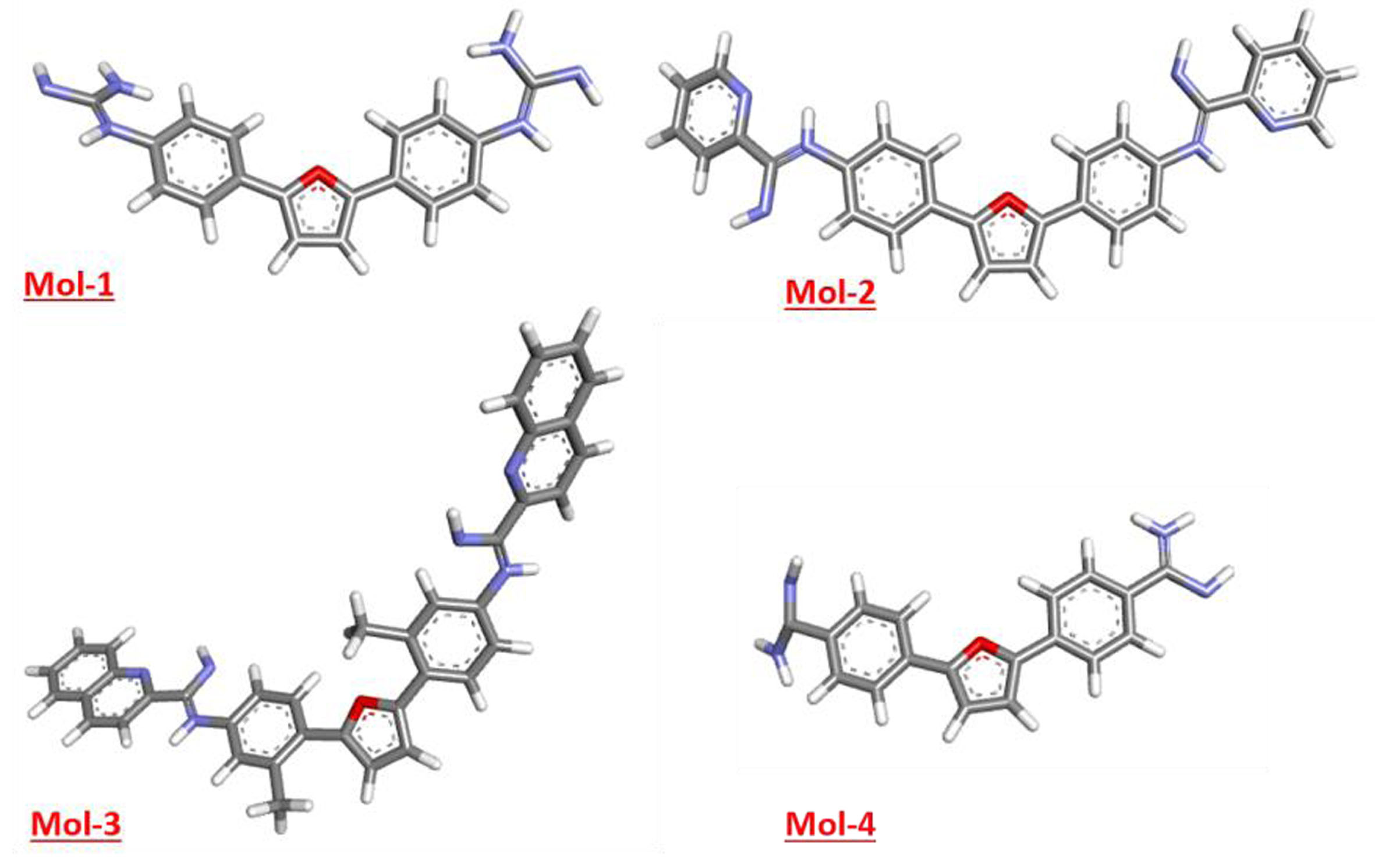
Figure showing optimized structures of the selected ligands.
Prior to docking, the lead molecules’ geometry optimization is essential to minimize the repulsion between the closest bonded atoms, between their angles, and between their dihedrals so that the lead molecules would achieve a most stable physical state that corresponds to the lowest energy [20, 40]. Figure 2 below displays the chosen ligands’ optimal geometries. The results obtained by the simulation studies are summarized in Table 1 (for electronic properties). Figure 3 shown below, represents the HOMO-LUMO orbitals for the selected ligands. Table 3 shows the HOMO-LUMO gap (band gap), further, various other parameters such as Band Gap (ΔE), Electron Affinity (EA), Ionization Potential (IP), Electronegativity (μ), and Chemical Potential (χ), were also calculated.
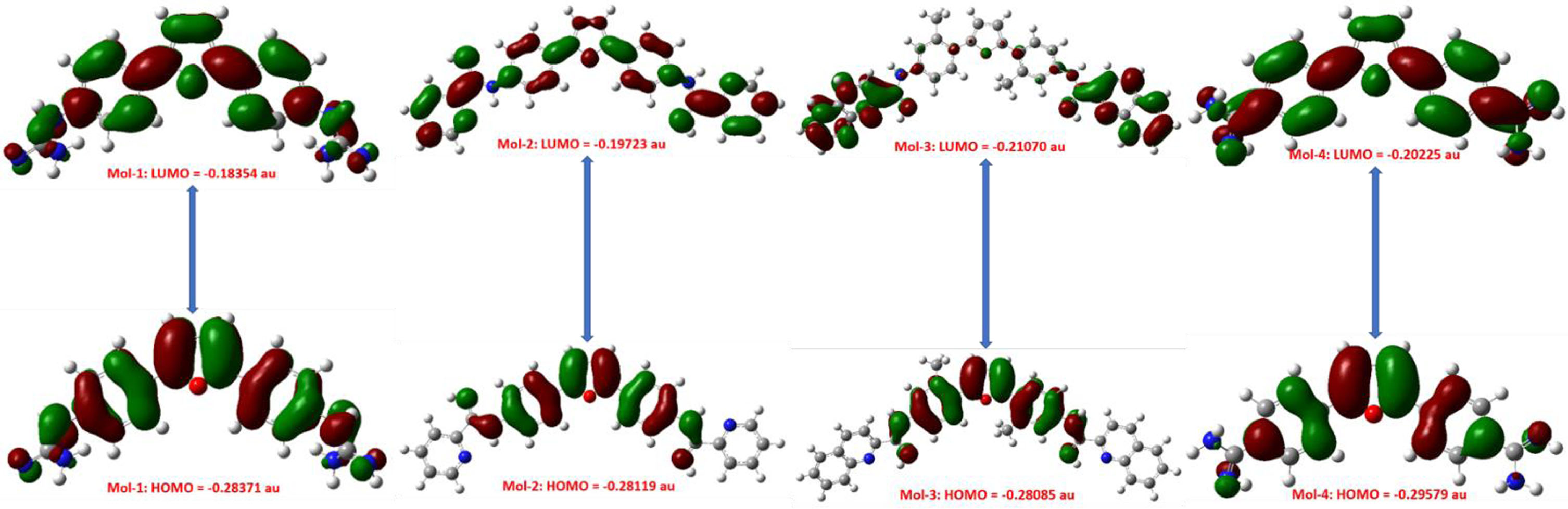
Figure represents the HOMO-LUMO orbitals for the selected ligands.
Table representing the electronic properties of the ligands without and with interaction from water molecules
Table representing various docking results obtained for 195D sequence
Following table represents the donor and the acceptor amino acid residues involved in the formation of hydrogen bond between the DNA and ligand atoms
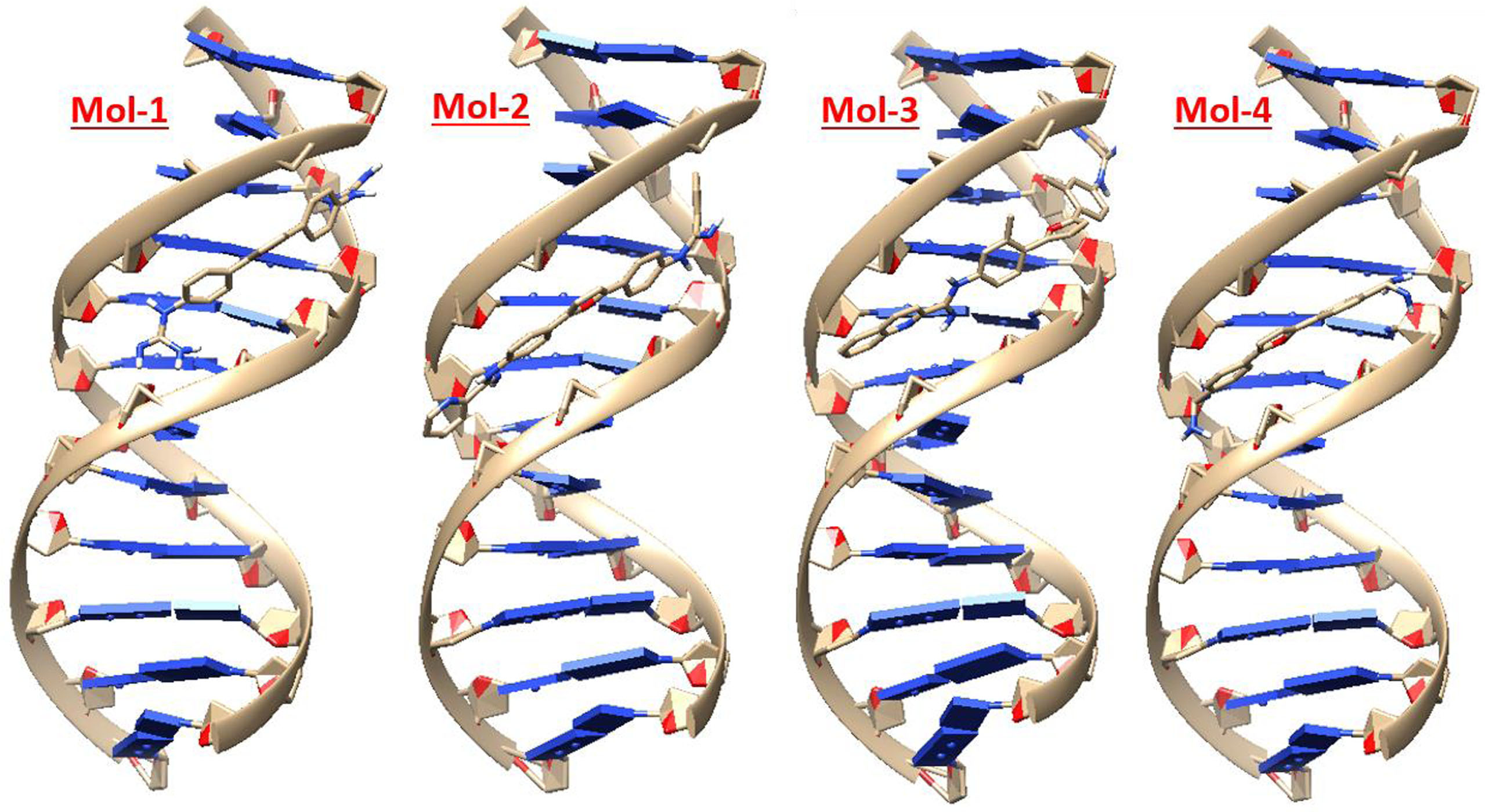
Figure showing best docked posed complexes for 195D in 3D.
Prior understanding of a therapeutic drug’s active binding site is quite crucial for determining how that drug interacts with duplex DNA. Henceforth, to intensely understand the preferred site for docking and the alignment of the drug molecule inside the DNA groove, molecular docking approach was used. From literature, derivatives of di-cationic (2,5-diaryl furans) were selected to assess for their potency as DNA minor groove binders. The four hypnotized ligands (mol-1, mol-2, mol-3 & mol-4) theoretically possessing antimicrobial tendencies by DNA minor grove binding were docked to 195D in quest of top docked posed complex. The best docked poses of all the proposed ligands in Fig. 3 clearly indicate that the proposed ligands could fit easily into the two base pairs, especially the A–T residues of minor groove of the DNA. The docking outcomes of all the ligands with 195D DNA sequences are listed below in Table 2. The molecular docking calculations exposed that 195D formed the most stable complex with third ligand (mol-3) with binding energy of –14.52 kcal/mol. 195D also formed its best docked posed complex with second ligand (mol-2) having binding energy of –11.62 kcal/mol. The order of ligands binding energy with 195D sequence follows the order mol3 > mol2 > mol4 > mol1 with mol3 to form the most stable complex with DNA. The mol3 aligns itself at the outer surface of AT base pairs that portrays its enhanced fit into the minor groove of DNA. The interaction of mol-3 with DNA most likely involves hydrogen bonds and van der Waals interactions. The root mean square deviation (RMSD) of mol3 with 195D sequence was also observed to be lowest of value 5.165 Å. Based on our docking results it is revealed that the mol3 forms three hydrogen bond; DTB:18 with -NH2 of carboxamide, O atom of furan ring and DGB:16 and DGB:14 with nitrogen of quinolone ring. Additionally, there is Π-anion interaction between imine nitrogen with DAB:19 and DGA:10 in mol3. In case of mol1, five hydrogen bonds [one H bond, -NH2 of carboximide and DAB:19, 2 H bond between DTB:18 and –NH2 of carboxamide, one H bond between O atom of furan ring and DGB:16 and 2 H bond between –NH2 of carboxamide and DGB:16 & DCB:15. In case of mol2 and mol4 2 and 3 H bonds were seen respectively with added Π-anion interaction between DTB:18 and Imine nitrogen in case of mol4. All these findings implied that all the four proposed ligands bond with minor groove of DNA with mol1 and mol3 having best binding as visualized from discovery studio software [41].
Hydrogen Bonding Analysis
Following Figs. 5 6 denote the binding site and corresponding H-donor/acceptor clouded regions at those binding sites, respectively. The number of hydrogen bonds formed, the interacting species along with the length of the hydrogen bond is mentioned in Table 3.
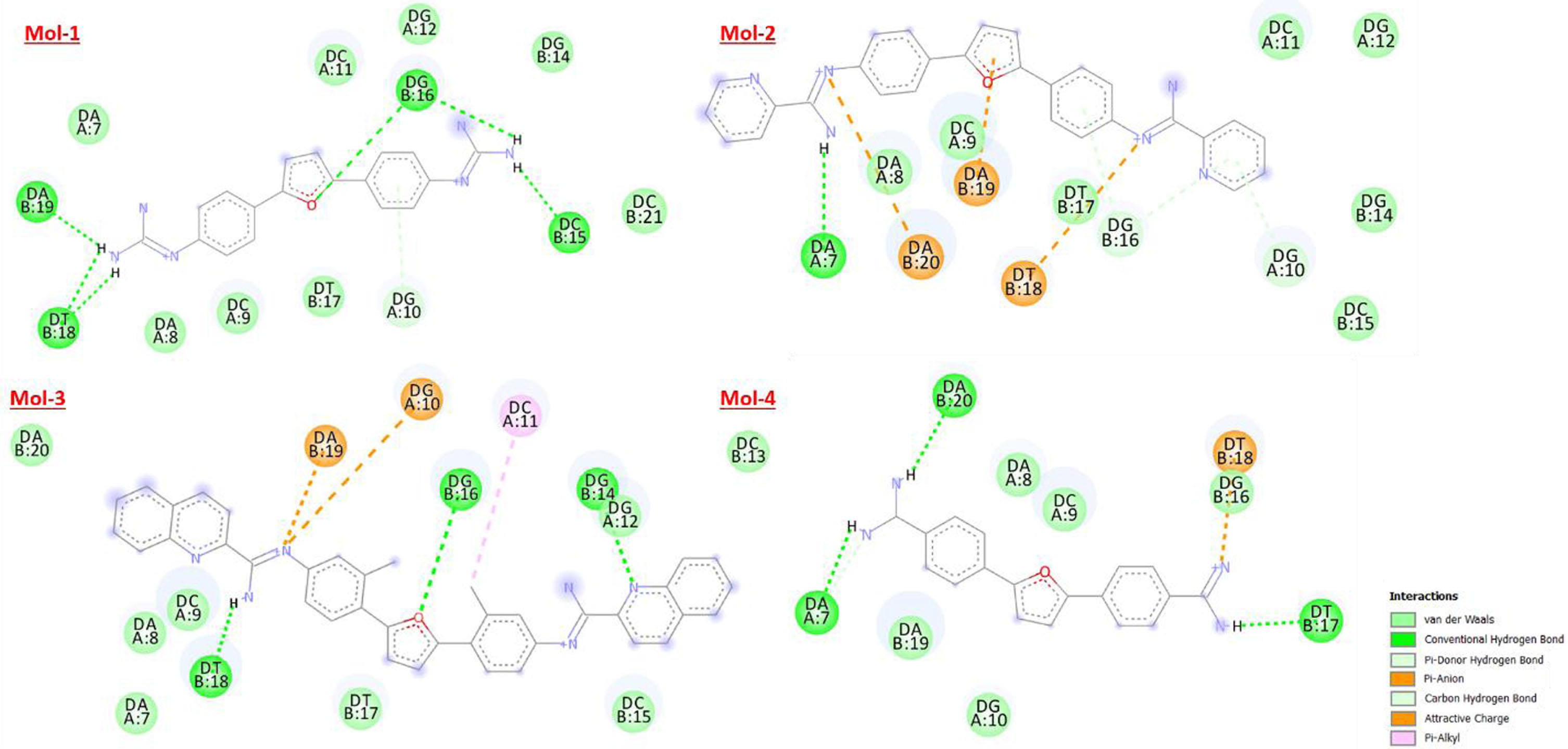
Figure showing the best docked posed complexes for 195D in 2D.
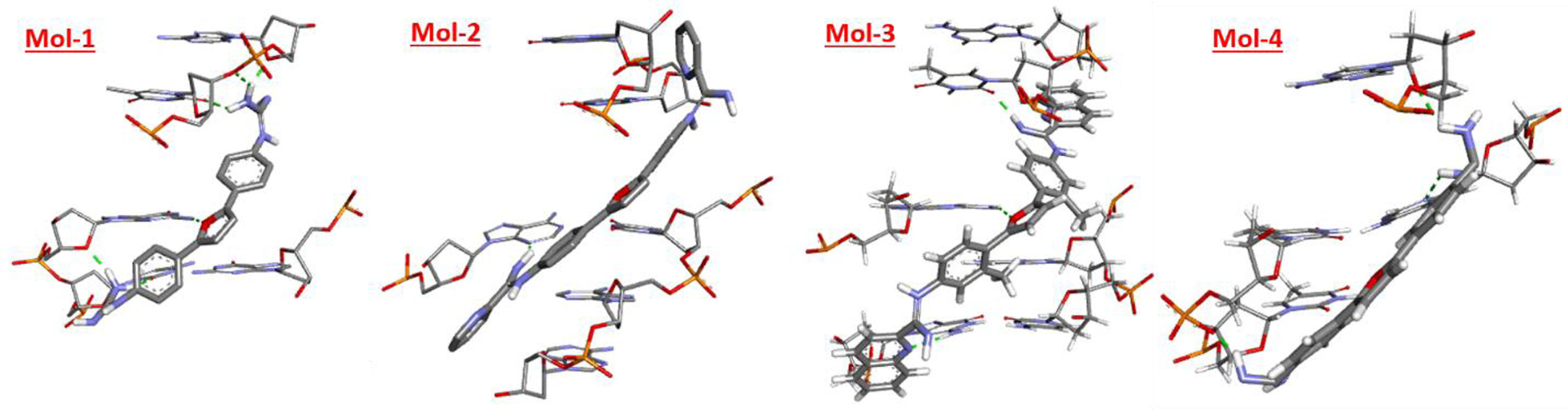
H-interaction sites for selected molecules with 195D.
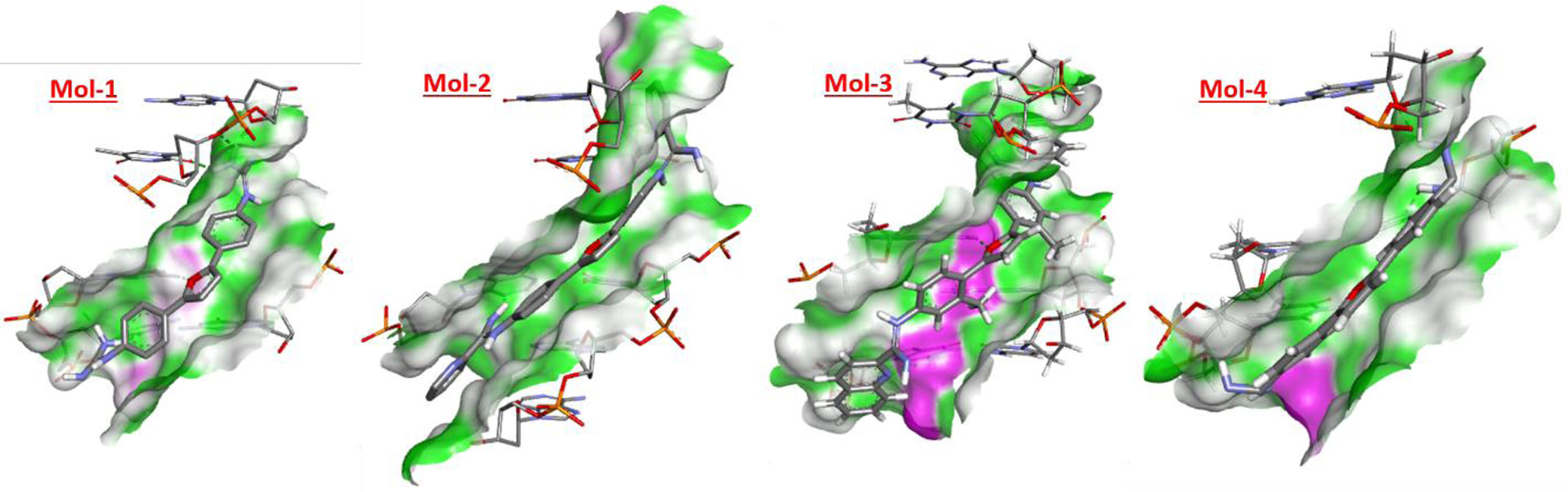
H-interaction sites for selected molecules with 195D.
Molecular dynamic simulation is used to study the structural stability of biomolecule under biological mimicked environment for a pre-defined time period. We analyzed the radius of gyration (Rg) to comprehend the dynamic stability and compactness of DNA-ligand complexes; which gave the average distance between the distinct base pair units and the averaged DNA center of mass. The collective Radius of Gyration variation data of 195D sequence DNA with four ligands i.e., mol-1, mol-2, mol-3 & mol-4 are shown in Fig. 7(a). The Rg data reveals that 195D sequences is mostly stable for its ligand complexes with avg. radiuses of gyration for 195D lie between 1.3 nm to 1.45 nm respectively. Mol-1-195D complex shows it remains highly compact during the entire simulation time period. In biomolecular interactions, hydrogen bonds are very important for determining the binding affinity and specificity. Figure 7(b) depicts the variations in the number of hydrogen bonds being formed during the simulation. We may see that mol-1 and mol-3 form four H-bonds during the dynamics on an average, whereas, mol-1 and mol-4 form three H-bonds on an average. This suggests a prominent H-bond interaction between DNA with mol-1 and mol-3, as already predicted by the docking analysis. We further calculated the RMSD depicting complex stability and structural equilibrium of the system are represented in Fig. 7(c). From Fig. 7(c) we can see that the deviations in DNA in complex-2 and complex-4 are maximum, which may be attributed to the steric clashes between the base pairs and the lead molecules, whereas complex-1 and complex-3 remain to be stable for the maximum duration. During the MD simulation, RMSF accounts for the variability in each pair of amino acid bases, as well as changes in the flexible areas of the nucleic acid. The low and high values of Root mean square fluctuations values can identify well-structured regions from loosely bonded regions in DNA strands respectively. The root mean square fluctuation (RMSF) plots of DNA nucleotides of four complexes are shown in Fig. 7(d). As fluctuation range of 1∼3 Å is tolerable for smaller protein during MD simulations. A least RMSF values of 0.05∼0.4 Å clearly seen in graph below suggest towards complex stability for both, 195D: mol-1 & 195D: mol-2.
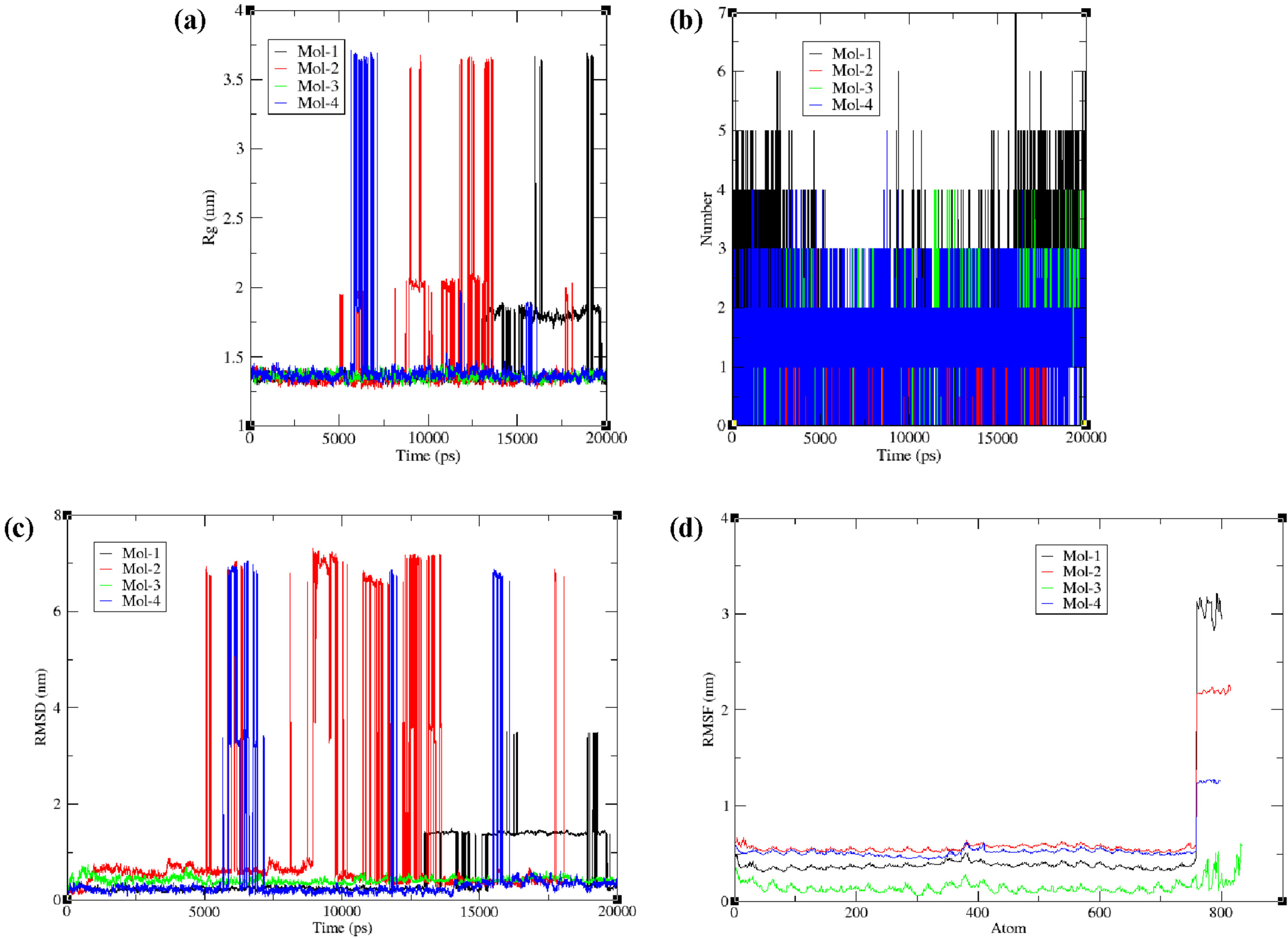
The molecular dynamics parameters (a) Rg, (b) H-bond, (c) RMSD, and (d) RMSF.
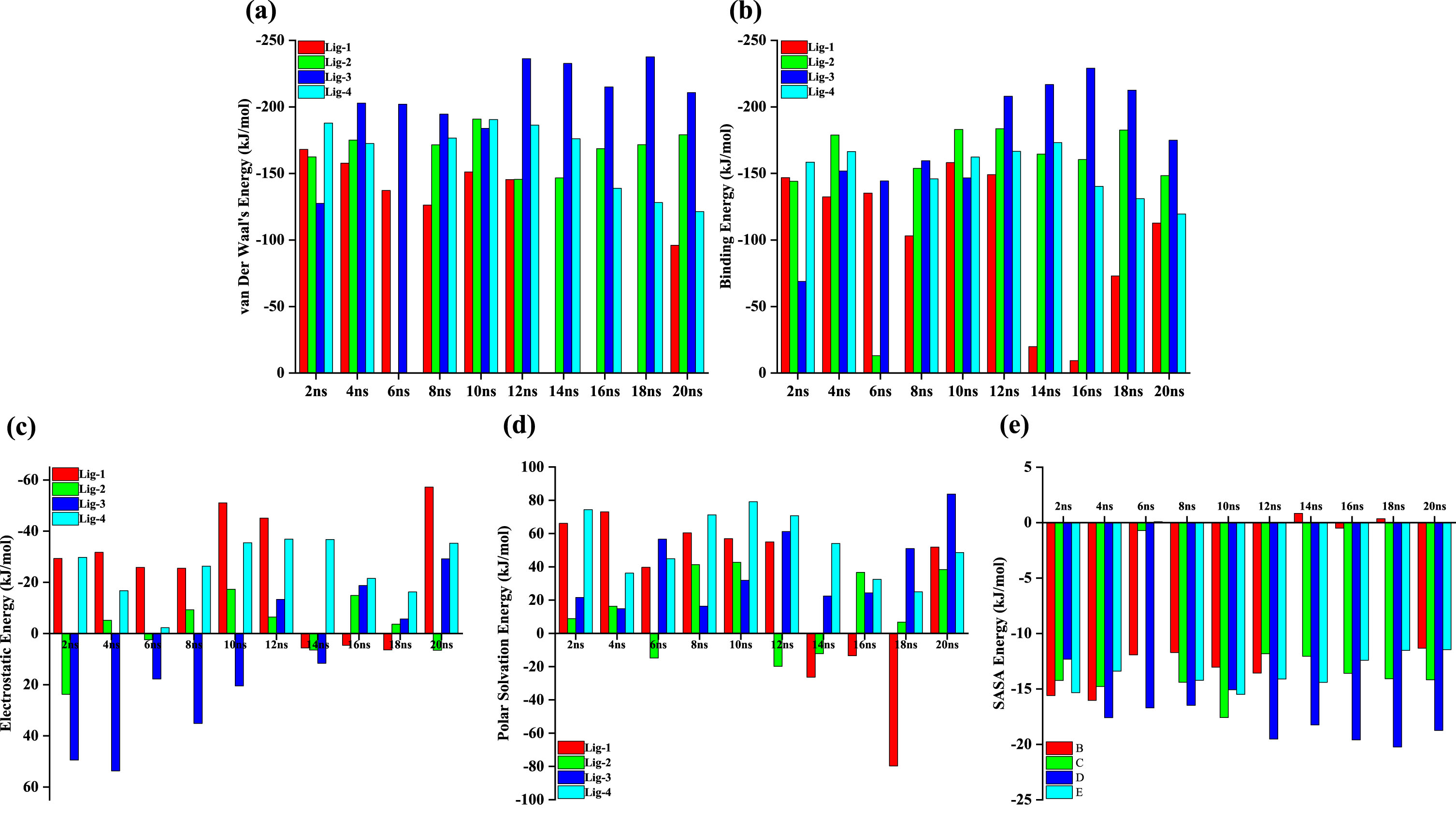
The free energy components (a) van Der Waal’s Energy (kJ/mol), (b) Binding Energy (kJ/mol), (c) Electrostatic Energy (kJ/mol), (d) Polar Solvation Energy (kJ/mol), and (e) SASA Energy (kJ/mol).
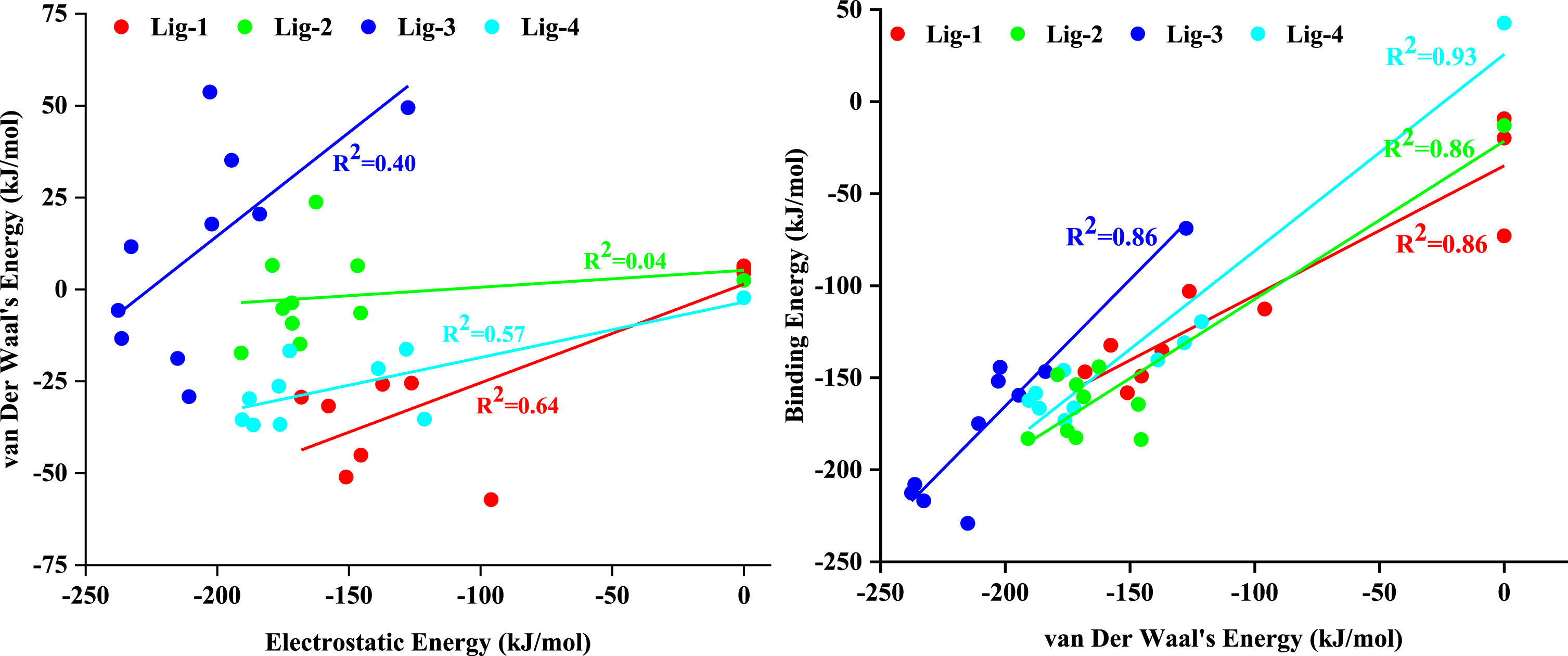
The correlation between free energy components (a) van Der Waal’s Energy (kJ/mol) with Electrostatic Energy (kJ/mol), and (b) van Der Waal’s Energy (kJ/mol) with Binding Energy (kJ/mol) for each DNA-ligand complex.
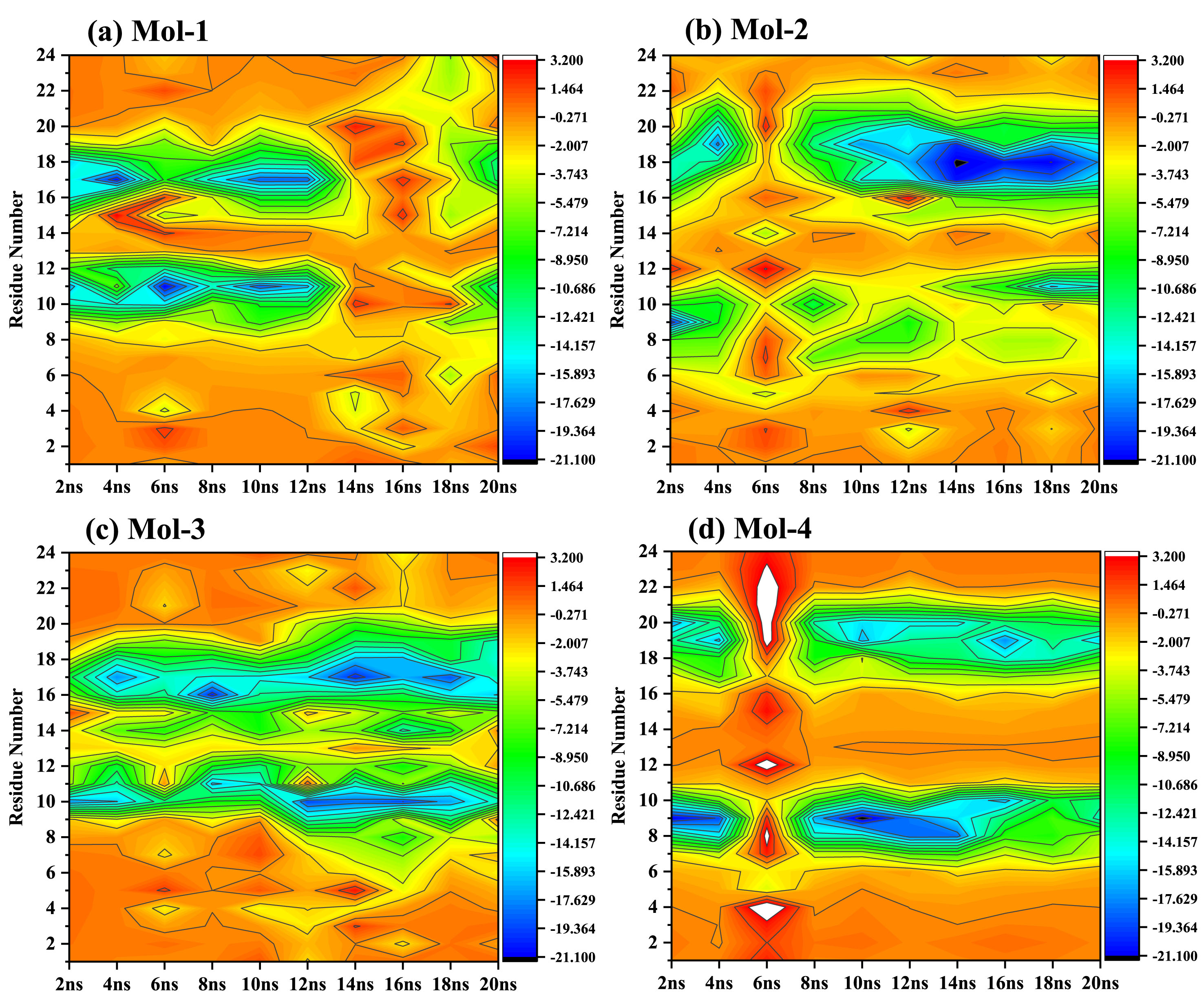
The heat map for per residue energy decomposition for each DNA-ligand complex.
In order to quantify the free energy components, we carried out free energy calculations for each of the DNA-ligand complex. We took 10 snapshots for each of the complex at an interval of 2 ns and read that frame to get the energetics. Table 4 lists out the free energy components, viz., van Der Waal’s energy, electrostatic energy, polar solvation energy, SASA energy, and binding energy, for each DNA-ligand complex. The relative variation of each of these parameters for each DNA-ligand complex can bee seen from Fig. 8, wherein we may see that DNA_lig-3 seems to have been consistently stable and held good energy values with the due course of the simulation. However, Table 4 presents us a mean value which indicates that all the four ligands possess good binding energies at each snapshot. We further derived correlation between free energy components, viz., van Der Waal’s energy with electrostatic energy, and van Der Waal’s energy with binding energy; for each DNA-ligand complex. Our observations reveal that there exists a good correlation between van Der Waal’s energy with binding energy (R2 = 0.86, R2 = 0.86, R2 = 0.86, and R2 = 0.93, respectively) than van Der Waal’s energy with electrostatic energy (R2 = 0.64, R2 = 0.04, R2 = 0.40, and R2 = 0.57, respectively). Further in order to look deeper into the per residue energy decomposition, we plotted a heat map as shown in Fig. 8, which reveals the energetically most stable and unstable regions of the DNA strands. We may see that residues around 8–12 and 16–20 are the ones which correspond to the AT-rich regions of the DNA while the remaining are the GC-rich regions. This also confirms that the selected ligands are AT-rich region selective. The heat map also indicates that lig-3 binds seemingly consistent and remains intact throughout the end of the simulation.
The free energy components (a) van Der Waal’s Energy (vDW, kJ/mol), (b) Electrostatic Energy (Ele, kJ/mol), (c) Polar Solvation Energy (Polar, kJ/mol), (d) SASA Energy (SASA, kJ/mol), and (e) Binding Energy (Bind, kJ/mol), for each DNA-ligand complexes
The free energy components (a) van Der Waal’s Energy (vDW, kJ/mol), (b) Electrostatic Energy (Ele, kJ/mol), (c) Polar Solvation Energy (Polar, kJ/mol), (d) SASA Energy (SASA, kJ/mol), and (e) Binding Energy (Bind, kJ/mol), for each DNA-ligand complexes
Bioinformatics/computational techniques have been frequently used in pharmaceutical industry to model drugs to their related biological targets. The complementing results of in-silico work other than biophysical or biological experimental data ease the future of any drug molecule. In the current research work, the interaction and stability of dicationic molecules in the vicinity of DNA has been studied. In short, the proposed ligands designed from literature were initially optimized through DFT studies followed by orientation assessment through molecular docking and finally subjected to molecular simulation for investigation of the stability of the DNA-ligand complex. The molecular docking data of all four ligands with 195D reveals their strong binding within the minor groove preferably at the A–T-rich sequence with ligand 3 depicting maximum binding affinity score –14.52 kcal/mol along with a minimum docking RMSD of 5.165. The various parameters of molecular simulation, free energy calculations and the heat map confirm the stability of 195D_mol-1 and 195D_mol-3 complex with the DNA through entire simulation period. Through our extensive investigation, we were able to locate most stable complex, 195D_mol-1 & 195D_mol-3. These encouraging outcomes from the in-silico studies of all the ligands direct towards their future pharmacological prospects and also provides deeper understanding of their mechanism. Moreover it also acts as initial simulated filtering step to complete chemical synthesis of a drug molecule and hence opens the gate for studying similar kind of molecule for improvement of existing drugs with higher potency in a cost effective manner.
Footnotes
Acknowledgments
Anwesh Pandey would like to acknowledge University Grants Commission, Govt. of India, New Delhi for the financial assistance during the course of this research work. Authors would also like to thank (Late) Dr. H.K. Srivastava, Associate Professor, Dept. of Medicinal Chemistry, NIPER, Guwahati, and Prof. Devesh Kumar for all the computational help & scientific discussions.
Conflict of interest
The authors declare no conflicts in their mutual interests, in any manner whatsoever.