
Abstract
Mesenchymal neoplasms of the kidney encompass a wide range of tumor types with heterogeneous clinical, histologic, and molecular features. Given their rarity relative to renal epithelial neoplasms, diagnosis may be challenging. In this review, the clinicopathologic and molecular characteristics of selected mesenchymal neoplasms of the adult kidney are discussed, including benign entities with diverse lines of differentiation (such as juxtaglomerular cell tumor, angiomyolipoma, and anastomosing hemangioma), sarcomas (synovial sarcoma, leiomyosarcoma, Ewing sarcoma), and tumors with variable biologic potential (solitary fibrous tumor). Mesenchymal lesions that may arise in peri-nephric soft tissue (including well differentiated / dedifferentiated liposarcoma and perinephric myxoid pseudotumor of fat) are also highlighted, given their potential to present as a ‘renal’ mass.
Keywords
INTRODUCTION
Mesenchymal neoplasms of the adult kidney are rare, but nevertheless comprise a wide range of tumor types with variable histologic appearances, lines of differentiation, and clinical behavior. This combination of rarity and diversity can make pathologic diagnosis challenging, whether evaluating a limited renal biopsy or a large nephrectomy specimen. In addition, as when occurring at other anatomic sites, mesenchymal tumors of the kidney may also share certain morphologic, immunophenotypic, and molecular findings, further complicating diagnosis. While some mesenchymal tumors of the kidney are specific to the site, such as juxtaglomerular cell tumor and renomedullary interstitial cell tumor (‘medullary fibroma’), the majority are entities that otherwise have a wide anatomic distribution. Examples of the latter include solitary fibrous tumor (SFT), synovial sarcoma, and Ewing sarcoma, tumors that can be harder to recognize when occurring as a renal primary. The precise anatomic site of mesenchymal neoplasms presenting as a “renal mass” can also vary, since such tumors may be based within the renal parenchyma (cortex or medulla), upper urinary tract, or peri-nephric soft tissue. In this review, the clinical, histological, immunohistochemical, and molecular features of selected mesenchymal neoplasms of the adult kidney are discussed, with an emphasis on those tumor types that are most common or diagnostically problematic. Mesenchymal neoplasms that are most frequent in the pediatric age group are not discussed but are covered in a separate review in this issue.
ANGIOMYOLIPOMA AND PECOMA
Angiomyolipoma (AML) is one of the most common benign mesenchymal neoplasms of the kidney. It is, as its name suggests, typically composed of adipocytes, smooth muscle, and blood vessels, a combination which initially led observers to regard it (incorrectly) as a hamartoma. AML is a member of the perivascular epithelioid cell tumor (PEComa) family, which is characterized by immunohistochemical expression of smooth muscle and melanocytic markers [1]. AML is most often sporadic, although a small subset is associated with tuberous sclerosis complex. In patients with this disorder, around 50% develop AML, which may be multiple and bilateral [2]. AML is usually well circumscribed and can arise in the renal capsule, cortex, or medulla. On imaging, a diagnosis of AML can oftentimes be presumed without biopsy when a renal mass has a significant fat component. Conversely, since AML with less fat (‘smooth muscle-predominant’ AML) has a wider radiologic differential diagnosis that includes malignancies, it is more frequently biopsied.
Histologically, AML commonly demonstrates smooth muscle, adipocytes, and abnormal thick-walled blood vessels, together imparting a ‘triphasic’ appearance, although the amount of each component can vary substantially (Fig. 1). AML, like other PEComas, co-expresses immunohistochemical markers of smooth muscle (smooth muscle actin, desmin, and caldesmon) and melanocytic (HMB45, melan-A, and MITF) differentiation, and it is invariably positive for at least one marker in each category. At the molecular level, AML commonly shows biallelic inactivation of TSC1 or TSC2 [3].

Angiomyolipoma. (A) Smooth muscle-predominant AML composed of spindle cells with eosinophilic and variably granular cytoplasm. (B) Fat-predominant AML can have a lipoma-like appearance. It demonstrates adipocytes and focal clusters of perivascular cells with clear-to-lightly eosinophilic cytoplasm.
Related tumors with more prominent epithelioid cytomorphology, and usually lacking a triphasic appearance, have long been termed ‘epithelioid AML’ [4]. Although the current WHO still recognizes epithelioid AML as a distinct variant, such tumors are often now diagnosed simply as ‘PEComa’, even though this is also the overarching term for this family of neoplasms [5]. Renal PEComa (epithelioid AML) usually demonstrates a predominant population of epithelioid tumor cells with abundant eosinophilic-to-clear granular cytoplasm. It can therefore mimic the histology of oncocytic renal epithelial neoplasms, such as oncocytoma and chromophobe renal cell carcinoma; however, PEComa is negative for PAX8. In addition to expression of smooth muscle and melanocytic markers, a small subset of PEComas driven by TFE3 gene fusions can exhibit nuclear expression of TFE3 [6]. In contrast with classic AML, PEComas (epithelioid AML) may have malignant potential. Malignant PEComas usually display mitotic activity, necrosis, and/or nuclear pleomorphism, and their recognition is clinically important since patients can benefit from mammalian target of rapamycin (MTOR) inhibitor therapy [7].
Of note, sclerosing PEComa is a distinctive variant that has a marked predilection for the retroperitoneum of female patients, and it may therefore present as a renal or peri-nephric mass [8]. Histologically, it is typically composed of cytologically bland epithelioid cells arranged in cords and trabeculae in a prominent sclerotic stroma. This variant is clinically indolent.
LEIOMYOMA AND LEIOMYOSARCOMA
Leiomyoma is a benign smooth muscle neoplasm that occurs rarely in the kidney. Grossly, it is often well circumscribed and grey/white. Microscopically, it is generally composed of fascicles of spindle cells with brightly eosinophilic cytoplasm and elongated ‘cigar-shaped’ nuclei. There may be areas of stromal hyalinization and degenerative changes. In a nephrectomy specimen, adequate sampling is important to assess for any mitotic activity, nuclear pleomorphism, or necrosis, which would raise concerns for leiomyosarcoma. By IHC, leiomyoma is typically diffusely positive for SMA, desmin, and/or caldesmon, while melanoma markers should be negative, helping to exclude AML / PEComa.
Leiomyosarcoma (LMS) – a malignant neoplasm with smooth muscle differentiation – occurs very rarely in the kidney but is the most common sarcoma to arise at this site [9]. On gross examination, LMS is often large (greater than 10.0 cm) with areas of extension beyond the kidney. Histologically, like leiomyoma, it usually comprises fascicles of spindle cells with eosinophilic cytoplasm and elongated blunt-ended nuclei. However, it also shows aggressive histologic features including mitotic activity, nuclear pleomorphism (and hyperchromatism), necrosis, and lymphovascular invasion. Most tumors are high-grade, exhibiting several of these features [9, 10]. By immunohistochemistry (IHC), LMS is also often positive for SMA, desmin, and caldesmon, while cytokeratin positivity can also be seen. Patients with LMS frequently develop metastases, often to the lungs, and have a poor prognosis [8]. The five-year survival is around 30% [11].
A subset of dedifferentiated liposarcomas demonstrate heterologous myogenic differentiation, such that this should be considered in the differential diagnosis of smooth muscle neoplasms, particularly when the patient has a large mass with involvement of retroperitoneal soft tissue. An adipocytic component may also be evident radiologically. When suspected, IHC for MDM2 and CDK4 (or molecular testing to assess for MDM2 amplification) can be helpful to exclude DDLPS (see also ‘Other diagnostic considerations’ below).
SOLITARY FIBROUS TUMOR
Solitary fibrous tumor (SFT) is a distinctive fibroblastic neoplasm that is genetically defined by a NAB2::STAT6 gene fusion. Histologically, it characteristically comprises spindle cells in a fibrous stroma with prominent branching staghorn-like blood vessels. The spindle cells typically have a haphazard distribution, imparting the so-called “patternless pattern”. In 2013, the oncogenic NAB2::STAT6 fusion was discovered [12, 13]. Soon thereafter, STAT6 IHC became established as a very sensitive and specific diagnostic marker [14]. It is typically used alongside CD34, which is also commonly positive, but much less specific. The biologic potential of SFT varies, ranging from tumors that are essentially benign to others that are highly malignant, and in recent years, risk assessment models have been developed to better predict clinical behavior [15, 16]. These scoring systems, adopted by the WHO classification [17], are based on patient age, tumor size, and mitotic activity, while an alternative four-variable model also includes necrosis. At the time of diagnosis, therefore, these criteria allow a given tumor to be designated as either low, intermediate, or high risk for distant metastasis, implying a five-year metastatic risk of 0%, 7–31%, or 51–100%, respectively [16].
ANASTOMOSING HEMANGIOMA
Anastomosing hemangioma is a benign vascular neoplasm with a predilection for the kidney, as well as perinephric soft tissue and other organs in the urinary tract [18]. It is usually solitary, although multifocal examples have been reported [19]. Microscopically, it tends to demonstrate a network of inter-connecting thin-walled vessels, lined by a single layer of endothelial cells with slightly protuberant or ‘hobnail’ nuclei. Additional histologic features which may support the diagnosis include the presence of intracytoplasmic hyaline globules and extramedullary hematopoiesis (usually most evident in the form of scattered megakaryocytes). Anastomosing hemangioma lacks the endothelial atypia and multilayering that are often present in angiosarcoma. Molecular studies have identified recurrent activating mutations in GNAQ, GNA14, and GNA11 [20].
RENOMEDULLARY INTERSTITIAL CELL TUMOUR
Renomedullary interstitial cell tumor (or ‘medullary fibroma’) is a very common benign mesenchymal neoplasm of the kidney that arises in the renal medulla. It is usually small (less than 0.6 cm) and encountered as an incidental finding in a nephrectomy specimen performed for another reason. If large enough to be apparent grossly, it is invariably described as a well circumscribed gray-white nodule involving the renal medulla. It consists of an unencapsulated proliferation of spindled and stellate cells in a loose stroma with peripheral entrapment of renal tubules. The cells have bland nuclei which lack any significant atypia or mitotic activity. There is variable positivity for CD34 and SMA, although IHC is rarely required to make the diagnosis.
MYOPERICYTOMA
Myopericytoma is a benign tumor with perivascular/pericytic differentiation that lies on a histologic spectrum with myofibroma. In the kidney, it is rare and usually discovered incidentally. Microscopically, myopericytoma often has a multinodular architecture and comprises spindle cells with palely eosinophilic cytoplasm and a ‘myoid’ appearance. There are typically prominent thin- and thick-walled blood vessels, around which the spindle cells may be oriented. Immunohistochemically, myopericytoma is usually positive for SMA and caldesmon. Desmin may be expressed focally in a subset. Genetically, myopericytoma/myofibroma harbors activating PDGFRB mutations [21, 22].
JUXTAGLOMERULAR CELL TUMOR
Juxtaglomerular cell tumor (JGCT) is a very rare neoplasm of the kidney that also represents an unusual cause of secondary hypertension due to its tendency to secrete renin, which in turn activates the renin-angiotensin-aldosterone system [23]. JGCT occurs mostly in adolescents and young adults and is slightly more common in females. Due to tumoral renin secretion, affected patients often present with hypertension, hyperaldosteronism, and hypokalemia, which may precede detection of the causative tumor by several years. On macroscopic examination, JGCT is usually small (less than 4.0 cm) and well circumscribed with a fibrous capsule. The histologic appearances are quite variable [24]. The neoplastic cells often have palely eosinophilic cytoplasm but may be polygonal or spindled. The growth pattern can be sheet-like or trabecular, and scattered cysts and tubules may be present (Fig. 2). There is usually no evident cytologic atypia or mitotic activity. In addition, JGCT typically demonstrates PAS-positive, diastase-resistant cytoplasmic granules and on electron microscopy there are rhomboid-shaped renin crystals. By IHC, there is positivity for CD34 and renin, with the latter characteristically showing diffuse granular cytoplasmic staining. SMA and CD117/KIT are also variably positive, while PAX8 and keratins are negative. Although the vast majority of JGCT are benign, malignant examples have been documented [24, 25].
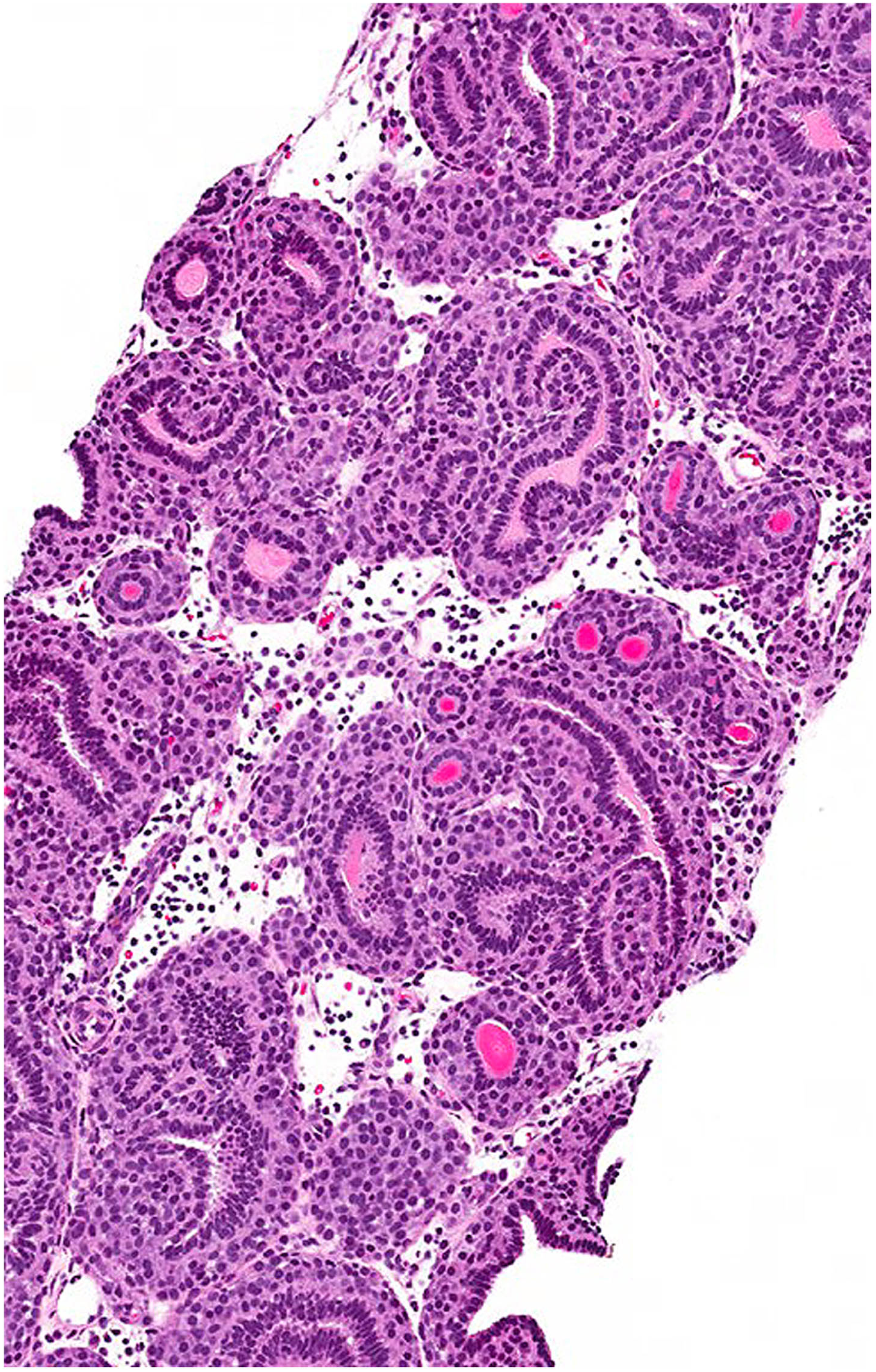
Juxtaglomerular cell tumor. This tumor type is specific to the kidney and often associated with hypertension due to renin production. This example shows tubules surrounded by a proliferation of cytologically uniform epithelioid cells with a glomoid appearance. (Courtesy of Dr. Michelle Downes, Toronto, Canada).
HEMANGIOBLASTOMA
Hemangioblastoma of the kidney is very rare, with less than 20 cases reported in the literature. Histologically, it resembles hemangioblastoma of the central nervous system, although in contrast, it does not appear to be associated with von Hippel-Lindau disease or harbor VHL alterations [26]. Microscopically, hemangioblastoma is circumscribed and typically composed of epithelioid/polygonal cells showing vacuolated clear-to-palely eosinophilic cytoplasm (Fig. 3). The cells usually have regular round-to-ovoid nuclei without significant atypia or mitotic activity. Characteristically, a prominent network of thin-walled vessels is also invariably present [27]. By IHC, hemangioblastoma tends to be positive for inhibin, S100-protein, and neuron-specific enolase (NSE), although the latter is not a very specific marker. Importantly, it may also express PAX8, and therefore be mistaken for renal epithelial neoplasms such as clear cell renal cell carcinoma [28]. Renal hemangioblastomas have not been reported to recur or metastasize.

Renal hemangioblastoma. The tumor is composed of epithelioid and polygonal cells that have clear cytoplasm and relatively uniform vesicular nuclei. The cells are associated with a network of thin-walled vessels, further helping to mimic the appearance of clear cell renal cell carcinoma.
EWING SARCOMA
Ewing sarcoma is an aggressive round cell sarcoma of bone and soft tissue that can rarely arise within the kidney [29–31]. It is molecularly defined by recurrent gene fusions between EWSR1 (or, occasionally, FUS) and members of the ETS transcription factor family (including FLI1, ERG, ETV4, and ETV1). The most common fusion is EWSR1::FLI1, generated by a t(11;22) translocation [32]. Until recently, ‘primitive neuroectodermal tumor’ (PNET) was employed as a relatively common synonym for Ewing sarcoma. However, since the term has also been used (rather confusingly) to refer to other unrelated round cell neoplasms, it is no longer recommended [32]. Renal Ewing sarcoma is seen most frequently in adolescents and young adults. Microscopically, as at other anatomic sites, it usually consists of primitive-appearing cells with uniform round nuclei and minimal cytoplasm. The cells are often arranged in sheets and occasional pseudorosette formation may be present (Fig. 4). By IHC, there is classically diffuse membranous expression of CD99, as well as diffuse nuclear expression of NKX2.2 [34]. A subset is positive for keratins. It should be distinguished from other round cell neoplasms, including CIC-rearranged sarcoma (see below), poorly differentiated synovial sarcoma (see below), blastema-predominant Wilms tumor, and lymphoma. Ewing sarcoma of the kidney has a poor prognosis. It is often locally advanced and frequently gives rise to metastases, most commonly to the lungs [35, 36].
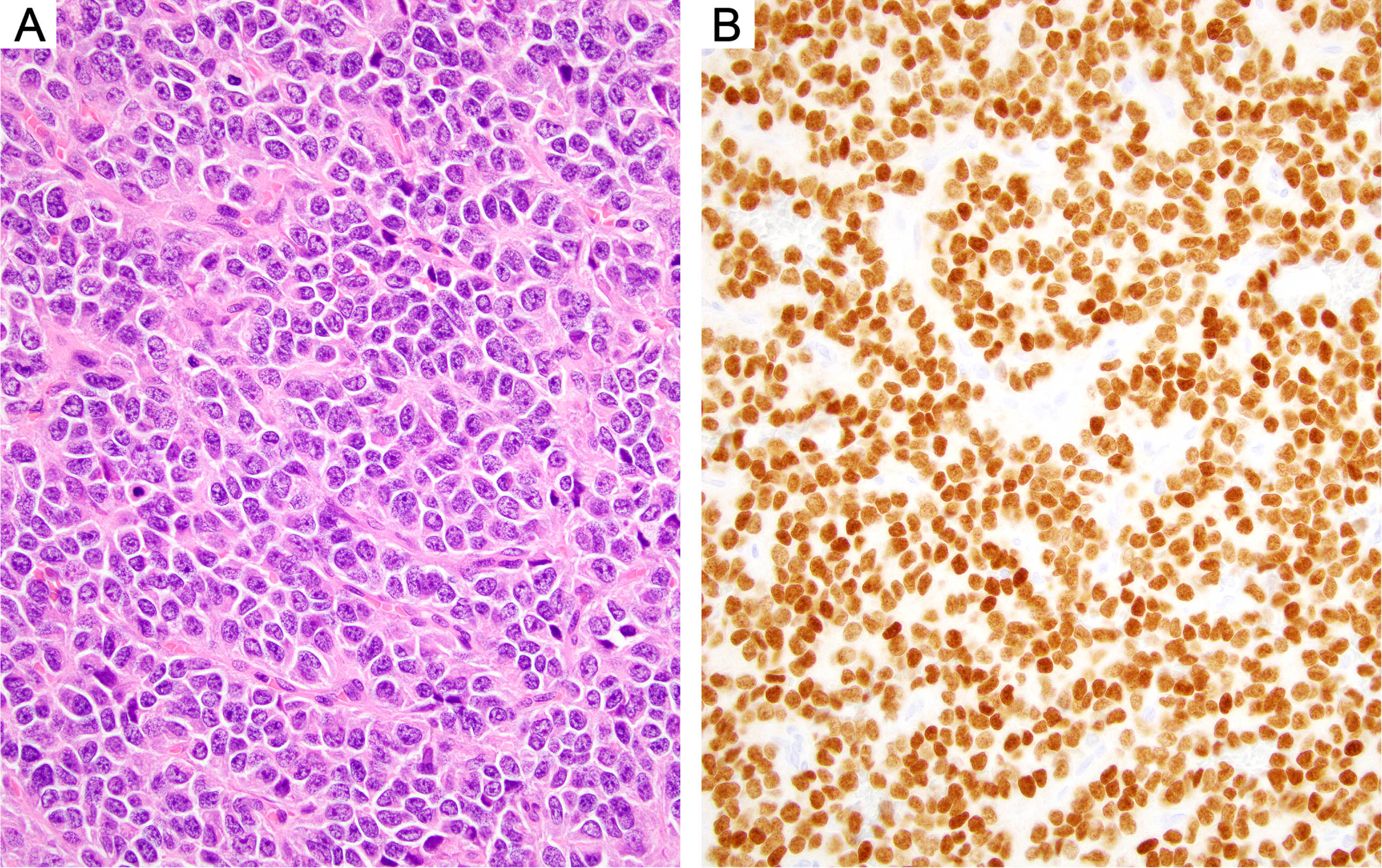
Ewing sarcoma. (A) The tumor comprises primitive-appearing small round blue cells with minimal cytoplasm. The cells are arranged in sheets and show focal pseudorosette formation. (B) Immunohistochemistry for NKX2.2 demonstrates strong and diffuse expression in the tumor cell nuclei.
CIC-REARRANGED SARCOMA
CIC-rearranged sarcoma is an aggressive round cell sarcoma that has been reported to occur in the kidney very rarely [37]. Prior to its recognition as a distinct entity, it was most likely classified among ‘Ewing-like’ sarcomas; however, unlike Ewing sarcoma, it tends to demonstrate more lobular architecture and be composed of cells with larger vesicular nuclei and prominent nucleoli [38]. There is usually only patchy positivity for CD99, as opposed to the more consistently diffuse membranous expression seen in Ewing sarcoma. Additional positive markers include ETV4 and WT1 [39]. Patients with CIC-rearranged sarcoma often receive chemotherapy with a Ewing sarcoma protocol, although responses are less frequent. It has a worse prognosis than Ewing sarcoma and a 53% two-year overall survival [37].
SYNOVIAL SARCOMA
Synovial sarcoma is a malignant mesenchymal tumor with variable keratin expression that is characterized by a recurrent t(X;18)(p11;q11) translocation. The latter generates an oncogenic fusion between SS18 and one of the SSX genes (SSX1, SSX2, or SSX4). Despite its name, it is unrelated to synovium, and may arise at various anatomic sites. In the kidney, it is typically circumscribed and unencapsulated. Histologically, it is classified as either monophasic, biphasic, or poorly differentiated. In the monophasic form, it is composed of cellular fascicles of spindle cells with minimal cytoplasm and relatively uniform overlapping nuclei. In biphasic examples, the spindle cells are also admixed with areas showing glandular differentiation. Poorly differentiated synovial sarcoma demonstrates round cell cytomorphology and increased mitotic activity; it therefore has a broader differential diagnosis that includes round cell sarcomas. Molecular confirmation is now much less commonly needed, given the reliability of IHC using two recently developed antibodies, SS18-SSX and SSX C-terminus. The former (95% sensitive, 100% specific) is directed against the junction of the fusion protein, while the latter (100% sensitive, 96% specific) detects a conserved region of the SSX1, SSX2, and SSX4 proteins [40, 41]. Other typically positive immunostains include EMA, keratins, and TLE1.
SARCOMA WITH MEIS1::NCOA2 FUSION
In 2018, primary renal sarcomas harboring MEIS1::NCOA2 fusions were described in two adult patients [42]. A subsequent case series has characterized additional examples of this emerging entity involving the kidney and other sites in the genitourinary tract [43]. Histologically, the tumor often has a nodular and variably cystic growth pattern with peripheral entrapment of renal tubules. It predominantly comprises monomorphic primitive-appearing spindle cells arranged in short fascicles with occasional foci of whorled architecture. Areas with a round cell morphology are commonly present, as are mitoses and necrosis. Since there is a non-specific IHC profile, including variable expression of TLE1, cyclin D1, and WT1, the diagnosis requires molecular confirmation. This tumor should be considered in the differential diagnosis with synovial sarcoma and BCOR-rearranged sarcoma.
OTHER DIAGNOSTIC CONSIDERATIONS
Peri-nephric soft tissue is a very common anatomic site for well differentiated liposarcoma / dedifferentiated liposarcoma (WDLPS / DDLPS), and it is not uncommon for this tumor to present as a ‘renal mass’ on imaging and be labeled as such when sampled by core needle biopsy [44]. WDLPS typically comprises variably sized adipocytes and cells with atypical hyperchromatic nuclei. DDLPS is usually a non-lipogenic and non-distinctive spindle cell neoplasm but can have a wide range of histologic appearances. Genetically, WDLPS and DDLPS are defined by amplification of part of chromosome 12 (12q14-15), encompassing MDM2 and CDK4. Consequently, IHC for MDM2 and CDK4 can be very helpful. If the histologic and IHC findings are equivocal, however, fluorescence in situ hybridization (FISH) is often used to confirm MDM2 amplification.
‘Perinephric myxoid pseudotumor of fat’ is a non-neoplastic / reactive lesion that may mimic WDLPS, both radiologically and histologically [45]. It occurs in patients who often have a history of renal disease (either neoplastic or non-neoplastic) [45, 46]. In addition, an association with urothelial carcinoma of the renal pelvis was also highlighted in a recent series [47]. Microscopically, it consists of mature adipose tissue with multifocally myxoid stroma containing bland spindle cells and a patchy chronic inflammatory cell infiltrate. Unlike WDLPS, cells with atypical hyperchromatic nuclei should not be present and the lesion lacks MDM2 amplification.
Finally, whenever faced with a cytologically atypical or malignant spindle cell neoplasm involving the kidney, the pathologist should always consider the possibility of sarcomatoid renal cell carcinoma (RCC) or sarcomatoid urothelial carcinoma. Of note, sarcomatoid RCC is not a specific diagnosis, but instead represents a form of dedifferentiation that can be seen in many RCC subtypes, the most common of which are clear cell and chromophobe. Overall, sarcomatoid features are present to some extent in around 5% of RCC [48, 49]. When assessing biopsy material, IHC is often essential if a conventional carcinoma component is not present. PAX8, a renal epithelial lineage marker, is expressed in around 70% of sarcomatoid RCC [50], though it can also rarely be positive in sarcomatoid urothelial carcinoma. If considering the latter, p63 / p40, GATA3, and keratins are useful additional IHC markers. A diagnosis of sarcomatoid carcinoma rather than sarcoma could prompt considerably different patient management, particularly if there is metastatic disease. Patients with sarcomatoid RCC could benefit from immune-checkpoint inhibitor therapy as studies have found increased expression of PD1/PDL1 in these tumors [49].
In summary, a wide variety of mesenchymal tumors can occur in the kidney in adult patients, and these demonstrate diverse histologic features and clinical behavior. Accurate diagnosis relies upon an awareness of the clinical and radiologic features, careful histologic assessment, and, in many cases, the judicious application of IHC and/or molecular tests. PEComa should be remembered as one of the most common mesenchymal tumors in the kidney, and one which can mimic the histology of renal epithelial tumors. In this regard, there should generally be a low threshold for using PAX8 whenever encountering a renal neoplasm that is potentially mesenchymal. Finally, one must also keep in mind the possibility of liposarcoma and other perinephric lesions (particularly when assessing biopsy material), since these may present as ‘renal’ masses clinically.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
The author reports no funding.
AUTHOR CONTRIBUTIONS
William Anderson wrote the manuscript; no data were generated.
CONFLICTS OF INTEREST
The author has no conflicts of interest to disclose.